Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Типы органических кислот. Факторы, определяющие кислотность. Примеры.
|
|
Способность органических соединений к ионизации изменяется в широких пределах. В водном растворе экспериментально возможно определить рКа лишь до значений -15 (рКа воды 15, 7). Для более слабых кислот определение рКа проводят в других растворителях. Например, в жидком аммиаке можно определять рКа до значения 33. Соотнести кислотность в воде и аммиаке можно, определив рКа для какого-либо соединения параллельно в этих двух средах, что позволяет осуществлять переход от одной шкалы к другой. Подход к оценке кислотности очень слабых СН-кислот - предложили полярографическую шкалу кислотности. Эта шкала охватывает кислоты, у которых кислотность изменяется в пределах 50 порядков. И все же полученные значения рКа для слабых СН-кислот являются весьма приблизительными.
Ввиду необъятно большого числа органических соединений невозможно для каждого из них иметь количественную оценку кислотных свойств. Действительно, константы рКа в различных растворителях установлены для относительно небольшого числа соединений и неизвестны для многих даже важных биологически активных веществ. Поэтому большое значение приобретает качественный подход к оценке кислотных свойств разных кислотных центров, который базируется на оценке стабильности сопряженного основания: (!) сила кислоты определяется стабильностью сопряженного основания (аниона), образующегося из этой кислоты. Чем стабильнее анион, тем сильнее кислота.
Другими словами, кислотность зависит от совокупности ряда факторов, обусловливающих стабильность аниона А-:
• электроотрицательности и поляризуемости элемента, отдающего протон;
• степени делокализации отрицательного заряда в анионе;
• способности аниона к сольватации, т. е. взаимодействию с молекулами растворителя.
Обычно на кислотность большинства веществ в растворе оказывают влияние одновременно несколько факторов, но в каждом конкретном случае один или несколько из них будут преобладающими. Ниже будет рассмотрена роль этих факторов в определении стабильности анионов (вначале без учета влияния среды). В отсутствие эффектов сольватации проявляется истинная (собственная) кислотность данного соединения. Собственная кислотность проявляется в газовой фазе, и в этом случае она определяется исключительно структурой соединения.
Природа атома в кислотном центре. Роль электроотрицательности и поляризуемости элемента в кислотном центре может быть наглядно продемонстрирована на примере кислот Брёнстеда с различными кислотными центрами, но с одинаковыми заместителями, в данном случае этильными радикалами:

По возрастанию кислотности кислоты Брёнстеда: 
В таком же порядке возрастает стабильность соответствующих анионов. Чем более электроотрицательным является элемент в кислотном центре, тем он более способен нести отрицательный заряд и соответственно тем стабильнее будет образующийся анион.
Поскольку электроотрицательность атома кислорода (3, 5) больше, чем атома азота (3, 0) и атома углерода (2, 5), то в таком же порядке будет уменьшаться стабильность соответствующих анионов. Сравниваемые элементы находятся в одном периоде, и их поляризуемость практически одинакова. Для элементов третьего и последующего периодов периодической системы основное влияние на стабильность аниона оказывает фактор поляризуемости. В приведенной выше группе кислот Брёнстеда в случае этантиол а атом серы больше по размеру и легче поляризуется, чем элементы второго периода (О, N, С) у других кислот. Отрицательный заряд на атоме серы делокализован в большей степени. Поэтому алкантиолят-ион стабильнее, чем соответствующий алкоксид-ион и т. д. В целом же SH-кислота будет сильнее ОН-, NH- и СН-кислот. Для представленной выборки соединений кислотность в газовой фазе и в растворе будет одинаковой в связи с тем, что сольватация близких по размеру анионов будет нивелирована. Тиолы, как более сильные кислоты, реагируют со щелочами, а также с оксидами, гидроксидами и солями тяжелых металлов. Со щелочными металлами тиолы образуют растворимые в воде соли, с тяжелыми металлами — нерастворимые:

Способность тиолов связывать ионы тяжелых металлов обусловила использование их в качестве противоядия при отравлениях соединениями мышьяка, ртути, хрома, висмута и других металлов, относящихся к тиоловым ядам.
Спирты, как слабые кислоты, практически не реагируют с гидроксидами металлов; значения рКа спиртов близки рКа воды, равной 15, 7. При взаимодействии этанола со щелочью равновесие сдвинуто в сторону исходного спирта и содержание этоксида натрия в реакционной смеси будет невелико. Однако спирты способны реагировать со щелочными металлами и сильными основаниями, такими, как гидриды или амиды металлов, литий- и магнийорганические реагенты:


Этиламин и пропан не проявляют заметных кислотных свойств. Тем не менее в других NH- и СН- кислотные свойства выражены гораздо сильнее, что обусловлено электроноакцепторным влиянием заместителей, связанных с кислотным центром.
Стабилизация аниона за счет сопряжения. При сравнительной оценке кислотности соединений, имеющих одинаковый элемент в кислотном центре, основным фактором, определяющим собственную кислотность, становится делокализация отрицательного заряда в анионе. Стабильность аниона значительно повышается, если имеется возможность делокализации отрицательного заряда по системе сопряженных связей. Характерным примером проявления действия этого фактора в группе ОН-кислот является повышение кислотности при переходе от спиртов к фенолам и к карбоновым кислотам.
Увеличение кислотности фенолов по сравнению с алифатическими спиртами объясняется большей стабильностью феноксид-иона, в котором отрицательный заряд делокализуется с участием атомов углерода бензольного кольца:

Повышенная по сравнению с фенолами кислотность карбоновых кислот обусловлена стабилизацией ацилат-ионов, в которых отрицательный заряд за счет р, π -сопряжения распределен поровну между двумя атомами кислорода:

Делокализация отрицательного заряда по системе сопряженных связей, приводящая к стабилизации аниона, приводит к увеличению кислотности и других типов кислот.
Влияние электронных эффектов заместителей, связанных с кислотным центром. Независимо от механизма передачи электронного влияния заместителя (индуктивного или мезомерного) в общем случае выполняется нижеприведенное правило: (!) Электроноакцепторные заместители способствуют делокализации отрицательного заряда, стабилизируют анион и тем самым увеличивают кислотность. Электронодонорные заместители, наоборот, понижают ее. Влияние на кислотность электроноакцепторных атомов галогенов наглядно иллюстрируется значениями рКа моно- и тригалогенозамещенных уксусных кислот. Наиболее сильный эффект оказывает самый электроотрицательный элемент фтор:

Влияние заместителей особенно ярко проявляется в ряду замещенных фенолов. Электроноакцепторная нитрогруппа, например, дополнительно стабилизирует образующийся анион, что приводит к увеличению кислотности n-нитрофенола (рКа 7, 1) по сравнению с незамещенным фенолом (рКа 10). Наличие в бензольном кольце трех нитрогрупп приводит к тому, что 2, 4, 6-тринитрофенол (пикриновая кислота) становится уже очень сильной кислотой (рКа 0, 8), сравнимой с минеральными кислотами. Электронодонорные метальная и аминогруппы дестабилизируют феноксид-ионы и уменьшают кислотность n-метилфенола (рКа 10, 1) и n-аминофенола (рКа 10, 5):

В ароматических кислотах влияние заместителей, находящихся в мета- и пара-положениях бензольного кольца, подчиняется общему правилу: электроноакцепторные — увеличивают кислотность, электронодонорные — уменьшают. Поведение орто-замещенных кислот часто бывает аномальным. Как правило, орто-замещенные бензойные кислоты сильнее соответствующих пара-изомеров, независимо от того, является ли заместитель донором или акцептором. Такое влияние заместителя называют орто-эффектом. Иногда орто-эффект имеет вполне очевидное объяснение. Например, n-гидроксибензойная кислота (рKa 4, 58) слабее бензойной (рKa 4, 19), как и ожидалось исходя из влияния на кислотность электронодонорной ОН-группы. Однако салициловая (о-гидроксибензойная) кислота гораздо сильнее (рКа 2, 98), поскольку в стабилизацию образующегося из этой кислоты салицилат-иона вносит вклад внутримолекулярная водородная связь, что и приводит к увеличению кислотности именно этого изомера:


Эффект сольватации. Влияние сольватации может быть очень значительным. Почти во всех случаях кислотно-основных взаимодействий можно считать, что исходные нейтральные молекулы и образующиеся ионы сольватируются по-разному. Стабильность аниона существенно зависит от его сольватации в растворе. Взаимодействие между растворителем и ионом может быть различным по своей природе — электростатическим, координационным (в том числе и за счет водородных связей), гидрофобным. При сольватации иона происходит перераспределение заряда с участием окружающих его молекул растворителя. Как правило, сольватация ионов в полярных растворителях тем сильнее, чем более полярен растворитель. Поскольку всю совокупность взаимодействий между ионом и окружающей его средой учесть чрезвычайно трудно, то обычно пользуются эмпирическим правилом: (!) чем меньше размер иона и чем больше локализован в нем заряд, тем он лучше сольватируется.
Трудным для интерпретации является вопрос о соотношении между кислотностью соединений в водной среде и газовой фазе. Развитие методов ионного циклотронного резонанса и масс-спектрометрии высокого давления обеспечило возможность достаточно точных определений термодинамических равновесий в газовой фазе. Так, было установлено, что по силе кислотности уксусная кислота и фенол в газовой фазе близки, тогда как в воде рКа этих соединений за счет эффекта гидратации анионов различаются на пять порядков. Полагают, что делокализация отрицательного заряда по бензольному кольцу в фенолят-ионе снижает его способность к образованию водородных связей с водой. Собственная кислотность алифатических спиртов в газовой фазе возрастает с увеличением длины и разветвленности алкильного радикала:

Алкильные группы могут участвовать в делокализации отрицательного заряда, потому что атом кислорода, несущий отрицательный заряд, является электронодонором по отношению к алкильным группам. В водном растворе приведенный выше порядок увеличения кислотных свойств данных спиртов меняется на противоположный. Такое обращение ряда кислотных свойств в воде по сравнению с газовой фазой объясняется лучшей гидратацией небольшого по размеру этоксид-иона и более слабой гидратацией объемного трет-бутоксид-иона. В газовой фазе спирты — более сильные кислоты, чем вода; в водном растворе — более слабые кислоты, чем вода:
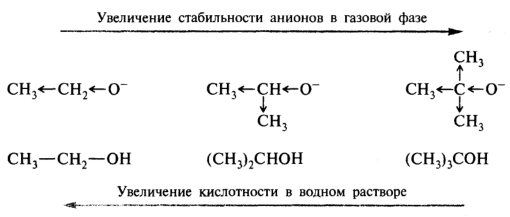
Таким образом, эффект сольватации может оказать более сильное воздействие, чем электронные эффекты заместителей. При прогнозировании преобладающего влияния можно руководствоваться в основном предельными случаями:
• в молекулах с резко различающимися по размеру углеводородными радикалами энергетический вклад сольватации больше, чем электронных эффектов. В растворе по сравнению с газовой фазой идет обращение ряда кислотности;
• в молекулах, имеющих близкие по размерам углеводородные радикалы (фенолы, ароматические кислоты и т. д.), энергетический вклад эффекта сольватации меньше, чем электронных эффектов. Порядок изменения кислотности в газовой фазе и водной среде совпадает.
|
|