Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Атеросклероз.
|
|
Термін “атеросклероз” походить від грецьких слів athere – кашка і sklerosis – твердий. У 1883 році J.Lobstein об’єднав декілька захворювань подібних за морфологічними змінами судинної стінки під терміном “атеросклероз”. Тепер в цій групі захворювань розрізняють 3 основні форми: атеросклероз, склероз середньої оболонки (артеріосклероз), і артеріолосклероз.
Атеросклероз – це хронічне вогнищеве ураження артерій, яке характеризується нагромадженням у внутрішній стінці артерій апопротеїн-В-вмісних ліпопротеїнів і холестерину, реактивним розростанням сполучної тканини з утворенням фіброзних бляшок, їх розривом, звиразкуванням, тромбозом, кальцинозом. Цей термін запропонував E.J.Marchand в 1904 році. Склероз і кальциноз судин м’язевого типу перебігає більш сприятливо, при цьому спостерігається ущільнення стінки судин, що спричиняє порушення кровоплину у тих ділянках, які ці судини харчують.
Артеріолосклероз характеризується ураженням артеріол з просякненням стінки судин білковими речовинами, які поступають з плазми крові (гіаліноз), з подальшим звуженням і навіть закриттям просвіту судин у селезінці, нирках, головному мозку (тут цей процес локалізується найбільш часто).
Епідеміологія. Атеросклероз частіше зустрічається у осіб, старших 45-50 років, одначе ранні атеросклеротичні зміни можна знайти в 30-35-річному і навіть у 20-25-річному віці й ще раніше. У чоловіків атеросклероз розвивається на 8-10 років раніше, ніж у жінок. Серед міського населення він спостерігається частіше, ніж серед сільських жителів.
Атеросклероз, і зв’язані з ним ускладнення (ІХС, АГ, інфаркт міокарда, мозковий інсульт), є основною причиною захворюваності і смертності населення. В Україні питома вага смертності внаслідок захворювань серцево-судинної системи складає 60%. В той же час у США, завдяки впровадженню в практику національних програм по боротьбі з атеросклерозом та артеріальною гіпертензією, за останні 20 років вдалося знизити смертність внаслідок зв’язаних з атеросклерозом захворювань на 30%. Це доказує реальність і перспективність модифікації стилю житття в масштабах популяції. На жаль, у нашій країні поки що такої тенденції ще не спостерігається.
Етіологія, патоморфологія і патогенез. Клінічні і біохімічні ознаки, які зустрічаються значно частіше серед осіб, у яких розвивається атеросклероз, ніж у популяції в цілому, отримали назву факторів ризику атеросклерозу. Значення концепції факторів ризику ровитку атеросклерозу є дуже важливою для практичної кардіології. Усьому світу відоме Фремінгемське дослідження, почате у США в 1947 році у невеликому містечку Фремінгемі, яке триває й досі. Саме в цьому дослідженні вперше, із великою долею ймовірності, доведено значення корекції факторів ризику атеросклерозу для смертності всієї популяції. Найбільш значимими факторами ризику атеросклерозу є гіперліпідемія (підвищення вмісту в крові рівня загального холестерину і триацилгліцеринів), артеріальна гіпертензія, паління, цукровий діабет, ожиріння. Доведено, що корекція цих, так званих модифікованих факторів ризику знижує ймовірність розвитку атеросклерозу.
Роль порушення обміну ліпідів. Поширеність розладів обміну ліпідів (дисліпідемій) дуже велика, до того ж вона тісно корелює з частотою атеросклерозу та ІХС. Так, серед чоловіків 40-50 років, мешканців економічно розвинутих країн Заходу, підвищення рівня загального холестерину в плазмі крові має місце у 45-57% випадків.
Основними ліпідами плазми крові є вільні, тобто неестерифіковані жирні кислоти, триацилгліцерини, фосфоліпіди та ефіри холестерину. Ліпіди в крові перебувають у зв’язку з білками, утворюючи комплекси, названі ліпопротеїдами. Неестерифіковані жирні кислоти зв’язані з альбумінами, всі інші ліпіди – з α - і β -глобулінами. Ліпопротеїди по суті є транспортною формою ліпідів, причому вони здійснюють транспорт ліпідів, як екзогенного (харчового) походження, так і ліпідів, які синтезуються печінкою і стінкою тонкої кишки (тобто ендогенних ліпідів). Окремі ліпопротеїди (так звані ліпопротеїди високої густини – ЛПВГ) захоплюють надмір холестерину з клітин периферійних тканин і повертають його назад у печінку для окислення в жовчні кислоти і виведення з жовчю.
Білки ліпопротеїдних комплексів отримали назву апопротеїнів (або просто “апо”). Плазмові ліпопротеїди складаються з ядра, в якому містяться триацилгліцерини і ефіри холестерину та оболонки, яка складається з фосфоліпідів, неетерифікованого холестерину і білка апопротеїну. До плазменних ліпопротеїдів відносяться хіломікрони, ліпопротеїди дуже низької густини (ЛПДНГ), ліпопротеїди низької густини (ЛПНГ), ліпопротеїди високої густини (ЛПВГ).
 |
Рис. 1. Структура ліпопротеїна.
В нормі вміст холестерину плазми крові не повинен перевищувати 5, 2 ммоль/л, триацилгліцеринів – 2, 3 ммоль/л, β -ліпопротеїдів – 6000 ммоль/л. Межовий рівень основних ліпідів крові: загального холестерину – 5, 2-6, 2 ммол/л, триацилгліцеринів – 2, 3-4, 5 ммоль/л. Підвищений рівень основних ліпідів крові: загального холестерину – > 6, 3 ммол/л, триацилгліцеринів – > 4, 5 ммоль/л.
Хіломікрони – утворюються в кишковій стінці в процесі всмоктування харчового жиру, вони призначені для транспорту екзогенних триаццилгліцеринів до місць утилізації (серцевий і скелетні м’язи, молочні залози тощо). В білковій частині хіломікронів знайдені (в мізерних кількостях) апопротеїни всіх основних груп В-48, С, Е, А. Протягом доби хіломікрони переносять від 70 до 150 г екзогенних триацилгліцеринів.
Ліпопротеїди дуже низької густини (ЛПДНГ) синтезуються переважно в печінці і є транспортною формою ендогенних триацилгліцеринів. За добу вони переносять від 25 до 50 г синтезованих в печінці триацилгліцеринів. Білка в них більше, ніж в хіломікронах. Тому вони здатні переміщатись в електричному полі (хіломікрони завжди залишаються на старті, бо білка в них мало). Головним білком у ЛПДНГ є апо-В і апо-С.
Ліпопротеїди низької густини (ЛПНГ) складають половину всіх ліпопротеїдів плазми крові людини, вони є головною транспортною формою холестерину в кровоплині. На долю апо-В-100 в ЛПНГ припадає 95-98%, лише 2-5% складають апо-С (І, ІІІ) і апо-Е. ЛПНГ є кінцевим продуктом метаболічних перетворень ЛПДНГ, що відбувається в кровоплиніі. Склад ЛПНГ свідчить про те, що вони утворюються в ході подальшого гідролізу триацилгліцеринів в присутності ліпопротеїдліпази. Останнім часом з'явилися повідомлення про можливість прямої секреції часток ЛПНГ печінкою. Головною функцією ЛПНГ є транспортуання холестерину у периферійні органи і тканини, якщо останні по якійсь причині не забезпечують себе своїм синтезованим холестерином. Катаболізм ЛПНГ здійснюється в основному у печінці, решта – в клітинах периферійних тканин. Отже, саме з процесом катаболізму холестерину нерозривно зв’язано постачання периферійних тканин холестерином, необхідним для побудови мембран, синтезу стероїдних і статевих гормонів, утворення жовчевих кислот. Кожна клітина організму здатна синтезувати холестерин, одначе велику частину холестерину більшість периферійних клітин отримують з ЛПНГ, які транспортують холестерин з місця його основного синтезу – печінки і кишечника, де утворюється не менше 90% всього холестерину.
Ліпопротеїди високої густини (ЛПВГ) – це ліпід-білкові комплекси, які мають у своєму складі найбільше білка і фосфоліпідів, відповідно 45-55%, і 24-40%. Білкова частина представлена апопротеїнами групи А (А І, А ІІ), лише 5% із загаль-ної маси білка приходиться на апо-С. В клінічній практиці в якості критерія вмісту в крові ЛПВГ використовується величина холестерину ЛПВГ, або α -холестерину (на противагу b-холестерину ЛПНГ). Утворення ЛПВГ відбувається в гепатоцитах і ентероцитах, а також в руслі крові в процесі катаболізму хіломікронів і ЛПДНГ. ЛПВГ, на відміну від ЛПНГ, здійснюють зворотній транспорт холестерину – з периферійних тканин до печінки для подальшого окислення жовчних кислот. Властивістю зв’язувати холестерин володіє один із основних апопротеїнів ЛПВГ апо-А І.
ЛПВГ захищають ЛПНГ від їх модифікації, внаслідок якої ліпопротеїди втрачають притаманні немодифікованим ліпідам властивості і набувають антигенних властивостей. В цілому, в організмі людини вирішальне значення має співвідношення ЛПНГ/ЛПВГ. Якщо це співвідношення підвищене, то утворення модифікованих ЛПНГ відбувається швидше, якщо понижене – навпаки, повільніше.
Модифіковані ліпопротеїди утворюються в організмі (в кровоплині, міжклітинних просторах органів і тканин) з нормально синтезованих і секретованих в кров ліпопротеїдів. Причинами модифікації можуть бути: викид клітинами вільних радикалів і продуктів пероксидації ліпідів, підвищена концентрація деяких метаболітів (глюкоза!), ферментів різного спектру дії.
Транспорт холестерину у складі ЛПНГ в клітину здійснюється шляхом рецептор-опосередкованого ендоцитозу. З’ясовано, що ендотеліоцити і гладком’язеві клітини артерій, лімфоцити, фібробласти шкіри мають на своїй поверхні специфічні рецептори білкової природи, здатні зв’язувати ЛПНГ. Процес взаємодії ЛПНГ з рецептором характеризується високою чутливістю і специфічністю. Один рецептор зв’язує одну частку ЛПНГ, загальне число рецепторів на поверхні клітини при 37˚ С коливається в межах від 15000 до 70000. За допомогою рецепторопосередкованого захоплення за добу з плазми крові видаляється біля 1г холестерину.
По іншому відбувається захоплення модифікованих ЛПНГ. Це виконують, зокрема, моноцити-макрофаги. Вони здійснюють нерегульоване скавенджер-захоплення ЛПНГ з утворенням пінистої клітини.
Переважна більшість пінистих клітин гине. При цьому в інтиму “вливаються” ефіри холестерину, кристали моногідрату холестерину. Утворюються вогнищеві скупчення холестерину, створюються умови для розвитку спочатку ліпідних плям, а потім і атеросклеротичних бляшок.
Отже, пінисту клітину можна розглядати не лише як клітину-попередницю розвитку атеросклерозу, але й як клітину-супутника атеросклерозу.
Міграцію моноцитів-макрофагів в інтиму артерій кваліфікують як один із проявів захисної функції РЕС, скерованої на захоплення і видалення модифікованих ЛПНГ. Разом з тим, макрофаги, виконуюючи цю функцію, і перетворюючись у пінисті клітини, гинуть на “полі бою”, їх смерть можна розглядати як механізм, який “запускає” атеросклеротичний процес. Кажуть, що макрофаг і “товариш”, і “злодій” здорового організму. При руйнуванні моноцитів-макрофагів виділяється значна кількість біологічно активних речовин, в тому числі й цитокінів (IL-1a, IL-8, MCP-1- monocyte chemoattractive protein-1), які посилюють міграцію інших моноцитів та лімфоцитів у субендотеліальний простір. Виникає “зачароване” патологічне коло (постійна рециркуляція моноцитів–пінистих клітин за принципом “інтима «кров”).
Здатністю захоплювати модифіковані ліпопротеїди, окрім макрофагів, володіють ендотеліоцити артерій. Вони ж володіють також так званою ретроендоцитозною активністю, завдяки якій відбувається видалення надмірних кількостей захоплених ЛПНГ з клітини. В цьому велика заслуга є й ЛПВГ.
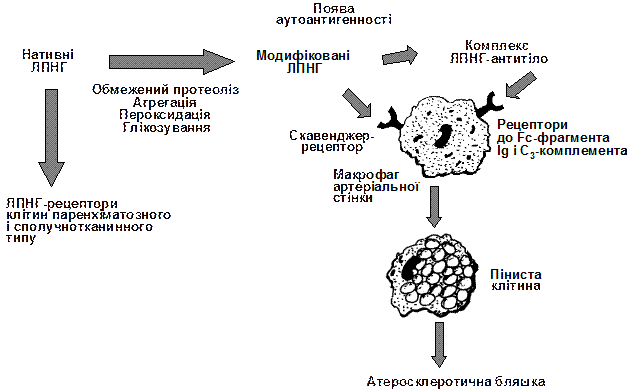 |
Рис. 3. Схема участі модифікованих ліпопротеїдів низької густини та імунних комплексів ліпопроїд-антитіло в атерогенезі
(за А.Н.Климовым, Н.Г.Никульчевой, 1998).
Існує точка зору про те, що ЛПНГ можуть проникати в інтиму артерій в обхід ендотеліальних клітин. Мова йде про концепцію міжклітинних проміжків, які в нормі дуже вузькі і непроникні для ЛПНГ. Проте у відповідь на гіперсекрецію катехоламінів, серотоніну, ангіотензину ІІ ці проміжки “розкриваються” в межах, достатніх для проникнення ЛПНГ.
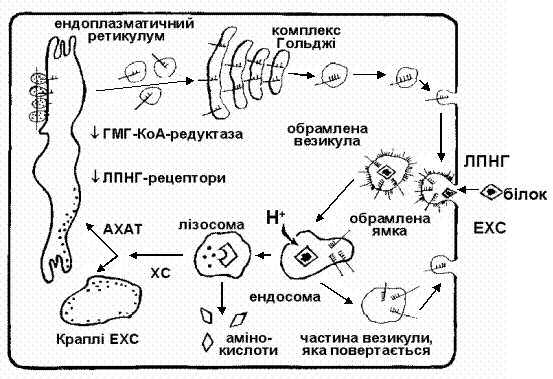 |
Рис. 2. Рецептор-обумовлене захоплення ліпопротеїнів. Захоплення ліпопротеїнів низької густини клітиною з участю специфічних рецепторів
(за M.Brown, J.Goldstein, 1984).
АХАТ – ацетил-КоА-ацетилтрансфераза (редуктаза);
ХС – холестерин; ЕХС – естерифікований холестерин.
Отже, перебіг атеросклерозу можна зобразити за схемою: ліпідна пляма→ фіброзна бляшка→ стабільна атеросклеротична бляшка→ нестабільна атеросклеротична бляшка (табл. 1).
| Таблиця 1. | ||
| Класифікація та послідовність розвитку атеросклеротичних змін у судинах (адаптовано за H. Stary, New York, 1994) | ||
| Типи ураження | Характеристика ураження | |
| І тип Початкові ураження | Нагромадження ліпопротеїдів у інтимі, ліпідів (ХС, ТГ) – у макрофагах. Виявляються мікроскопічними або гістохімічними методами. Структурні зміни відсутні. | |
| IІ тип Ліпідні плями | Нагромадження ліпопротеїдів у інтимі, ліпідів (ХС, ТГ) – у макрофагах та гладком’язевих клітинах. Видні неозброєним оком (плями). Структурні зміни відсутні. | |
| ІІа тип | Ліпідні плями – із схильністю до прогресування | |
| ІІ б тип | Ліпідні плями – несхильні до прогресування | |
| ІІІ тип Преатерома | Зміни типу ІІа + чисельні позаклітинні відкладання ліпідів. Мікроскопічно: ознаки тканинного пошкодження та дезорганізації | |
| ІV тип Атерома | Усі зміни ІІа і ІІІ типів + формування ліпідного ядра. Виражене порушення структурної цілості інтими. Порушення гемодинаміки. | |
| V тип Фіброатерома | Усі зміни ІІа, ІІІ, ІV типів + проліферація гладком’язевих клітин поверх ліпідного ядра, формування “покришки”. Є порушення гемодинаміки внаслідок звуження просвіту. | |
| VI тип Нестабільна атеросклеротична бляшка | VІa – розрив бляшки VІb – крововилив у бляшку VIc – тромбоз бляшки | Ускладнення атеросклеротичної бляшки: -інфаркт міокарда -мозковий інсульт -гангрена |
| VІІ тип Кальцифікація бляшки | Порушення гемодинаміки в залежності від локалізації | |
| VІІІ тип Фіброзна бляшка | Стабільне порушення кровоплину |
Ліпідні плями утворюються в різних ділянках артеріальної системи, але найшвидше в аорті, де їх знаходять уже в 10-річному віці (тоді вони займають 10%, а в 25 років і до 30-50% поверхні аорти). До 15-річного віку ліпідні плями появляються у вінцевих артеріях серця, а в 35-45 років вони є і в артеріях мозку. В ліпідних плямах переважають пінисті клітини, ефіри холестерин є переважно в клітин, хоч частково є й поза клітинами. Ліпідні плями не є перешкодою для циркуляції крові в судинах. Основним елементом атеросклеротичної бляшки є піниста клітина, руйнування якої спричиняє проліферативну реакцію з боку гладком’язевих клітин, які втрачають здатність скорочуватись; посилено синтезують сполучнотканинні білки (еластин, колаген). Фіброзна тканина, що утворюється з цих білків, у вигляді капсули оточує холестеринові вогнища, стараючись повністю їх ізолювати. Утворюється так звана фіброзна бляшка, в якій вміст ефірів холестерину в 20-26 разів, а неестерефікованого холестерину в 6-7 разів перевищує кількість стеринів в непошкоджених ділянках. Фіброзні бляшки збільшуються внаслідок крововиливів в них, фібриноїдного просякання, нагромадження в них ліпідів, пристінкового тромбозу. Останньому сприяють тріщини або розриви фіброзного покриття. Фіброзні бляшки часто звиразковуються, така поверхня стає місцем утворення пристінкового тромбу. Крім того така бляшка може бути щілиною виходу атеросклеротичного вмісту у кров’яне русло та емболії артерій мозку та інших органів. Ускладненням фіброзної бляшки є також її звапнення (атерокальциноз). Вогнища кальцинозу частіше зустрічаються в черевному відділі аорти і гирлах судин, що відходять від неї, у вінцевих артеріях серця.
Часто в одній і тій же артерії можна спостерігати різні стадії атеросклеротичних змін (ліпідні плями і фіброзні бляшки), що є результатом хвилеподібного перебігу атеросклеротичного процесу, який має фази прогресування, стабілізації і регресування. Фіброзні бляшки є перешкодою до кровоплину, вони можуть призводити до ішемії органа або тканин. З приводу можливості регресії атеросклерозу варте уваги наступне. Ліпідні плями, як початкові морфологічні прояви атеросклерозу, можуть зникати в будь-якому віці, якщо по якійсь причині відбулося значне і тривале зниження рівня холестерину в плазмі крові. Що ж торкається атеросклеротичних ураженнь на стадії фіброзної бляшки, то вони, по суті, регресії не піддаються. Одначе у фіброзних бляшках може відбуватись часткове або й повне зникнення ліпідів (деліпідація), в зв’язку з чим бляшка може стати більш плоскою. Це веде до поліпшення циркуляції крові через уражену судину. Таке явище носить назву часткової регресії. Важливу роль у видаленні ліпідів з атеросклеротичної бляшки відіграють макрофаги, які можуть захоплювати шляхом фагоцитозу навіть кристалічний холестерин. Для регресії атеросклерозу характерне поступове зникнення пінистих клітин, зменшення некротичної маси у серцевині ядра бляшки. Щоб домогтися часткової регресії, необхідно знизити загальний холестерин плазми до так званого ідеального рівня (3, 87-4, 13 ммоль/л) і підтримати цей рівень впродовж 1, 5-2 років. Хорошим передвісником деліпідації фіброзних бляшок є регресія шкірних ксантом.
Таким чином, на різних стадіях морфогенезу атеросклерозу, починаючи від проникнення ліпопротеїдів в артеріальну стінку, приймають участь п’ять типів клітин: ендотеліальні, гладко-м’язові, моноцити (макрофаги), тромбоцити, лімфоцити. Первинним субстратом, винним в утворенні пінистих клітин, є модифіковані апо-В-вмісні ліпопротеїди (апо-В-ЛПНГ). Саме вони є джерелом холестерину для утворення пінистої клітини і формування атеросклеротичної бляшки.
Отже, холестеринова концепція атеросклерозу, сформована М.М.Анічковим та С.С.Халатовим у 1913 році, витримала випробування часом. Еволюція цієї концепції може бути подана так: 1) без холестерину не може бути атеросклерозу (М.М.Анічков, С.С.Халатов 1913); 2) холестерин плазми крові корелює з небезпекою розвитку атеросклерозу (20-40-ві роки); 3) ліпопротеїди, як носії холестерину, відповідальні за розвиток атеросклерозу (40-50-ті роки); 4) ЛПНГ і ЛПДНГ є атерогенними, а ЛПВГ антиатерогенними і захищають організм від атеросклерозу (60-70-ті роки); 5) модифіковані ЛПНГ і ЛПДНГ в першу чергу відповідальні за розвиток атеросклерозу (80-90-ті роки).
Окрім холестеринової теорії атеросклерозу, яка в подальшому “трансформувалась” у теорію “модифікованих ліпопротеїдів низької густини”, широкого поширення набула також теорія “відповіді на пошкодження”. Основою її є концепція Р.Вірхова, яка тепер розвинута R.Ross (1976) та іншими вченими. Вона передбачає: 1) пошкодження ендотелію; 2) звільнення факторів, які викликають міграцію гладком’язевих клітин крізь внутрішю еластичну мембрану та їх проліферацію в інтимі, а також міграцію моноцитів та СD3-лімфоцитів (Т-клітин) периферійної крові крізь міжендотеліальні щілини з нагромадженням їх в субендотеліальному просторі; 3) синтез м’язовими клітинами колагену, еластину, протеогліканів; 4) внутрішньо- і позаклітинне нагромадження ліпідів; 5) утворення тромбу.
Основними причинами пошкодження ендотелію вважають: гіперліпідемію; підвищення стенічної напруги (механічний чинник), особливо у місцях біфуркації або розгалуження артеріальних стовбурів (стиснення ендотелію внаслідок напруги зсуву); вплив інфекції (віруси герпеса, Епштейна-Бара, цитомегаловіруси, хламідії, хеліобактерна інфекція тощо) за умови хронічного імунодефіциту, паління; вплив циркулюючих імунних комплексів, катехоламінів, оксиду вуглецю (паління), артеріальної гіпоксемії.
При пошкодженні ендотелію відбувається його десквамація з оголенням тромбогенного субендотеліального колагену з наступною адгезією і агрегацією тромбоцитів, вивільненням тромбоцитарного фактора росту, який стимулює міграцію і проліферацію гладком’язевих клітин у місцях пошкодження. Хемотаксичною властивістю володіє також b-тромбоглобулін пластинок та тромбоксан А2, який посилює агрегацію тромбоцитів і місцеву вазоконстрикцію.
У 1997 році теорія “відповіді на пошкодення” доповнена імунологічною концепцією (G.Wick, 1997), згідно з якою різноманітні стресові чинники, в тому числі і фактори ризика атеросклерозу, індукують пошкодження ендотелію і утворення “стресових білків” або “білків теплового шока” (Heat Shock Proteins, HSP). Експресію HSP особливо активно індукують: висока температура, вільні радикали, механічні чинники, цитокіни, важкі метали. HSP розцінюються імунною системою як чужерідні або “криптні” антигени, що дає початок справжній аутоімунній реакції. В атеросклеротичних бляшках знайдені цілі родини “білків теплового шоку” з 65 і 70 білків, а на ділянках, схильних до розвитку атеросклерозу – з 60 білків (відповідно, HSP65, HSP70, HSP60). У відповідь на експресію ендотелієм білків теплового шоку (HSP) виникає запальна і аутоімунна реакція, яка опосередкована, в основному, моноцитами і СD3-лімфоцитами (Т-клітини). На цьому етапі велике значення мають також адгезивні молекули, найважливішими з яких є: мікроцитарна адгезивна молекула (LAM), міжклітинна адгезивна молекула – intercellular adhesion molecule-1 (ICAM-1), судинно-клітинна адгезивна молекула – vascular cell adhesion molecule-1, які опосередковують, відповідно, лейкоцитарно-ендотеліальне зв’язування лейкоцитів (LAM), кооперацію гранулоцитів, моноцитів і лейкоцитів (ІСАМ-1), селективну інфільтрацію ділянки пошкодження моноцитами і лімфоцитами (VCAM-1). Активація міжлейкоцитарної кооперації супроводжується утворенням лейкотрієнів (В4), цитокінів-інтерлейкінів (IL-1, IL-2, IL-4, IL-8, g-інтерферона), туморнекротичного фактора, які посилюють проліферацію зрілих і молодих тимоцитів, стимулюють кілерну функцію лімфоцитів [CD56(NK)]. Міжпопуляційна кооперація лейкоцитів особливо активується за умов гіперхолестеринемії та модифікації ліпопротеїнів низької густини. За таких умов відбувається посилене усворення анти-HSP65-антитіл, які викликають лізис тих ендотеліальних клітин, які піддались впливу стресора. Доведено, що титр антитіл до білка теплового шоку HSP65 суттєво знижується у хворих після черезшкірної транслюмінальної коронарної ангіопластики (ЧТКА), а також після активної антихелікобактерної терапії та лікування хламідіазу. Ця концепція атеросклерозу тепер є головною.
Існують і інші концепції патогенезу атеросклерозу - моноклональна та концепція про роль гіпервітамінозу вітаміну Д в атерогенезі.
Згідно моноклональної теорії проліферація гладком’язових клітин і ріст атеросклеротичної бляшки здійснюється з однієї мутантної клітини, подібно до клітин доброякісної пухлини. Під впливом мутагенів (віруси, паління) частина гладком’язових клітин піддається мутації. Мова йде про “підпороговий неопластичний стан”. В такому стані клітини можуть існувати роками, але згодом під впливом гіпертензії та гіперхолестеринемії найбільш чутливі гладком’язові клітини починають з великою швидкістю проліферуватися, що веде до утворення бляшки з моноклональним набором клітин.
Гіперліпідемії. Гіперліпопротеїдемія (ГЛП) – найважливіший фактор ризику атеросклерозу. Розрізняють первинні і вторинні гіперліпідемії (табл. 2).
Коротка характеристика гіперліпідемій та можливі шляхи їх корекції.
Родинна гіперхіломікронемія – ГЛП 1-го типу – зустрічається порівняно рідко. Клінічні ознаки можуть проявитися в дитячому віці, іноді перші симптоми виявляються у дорослих. Характерне різке підвищення рівня триацилгліцеринів (від 4, 5 до 112, 9 ммоль/л, при нормі до 2, 3 ммоль/л), що відбувається за рахунок фракціі хіломікронів. При стоянні плазми такого хворого в холодильнику упродовж 24 години з’являється вершкоподібний пласт. Ознаками цього типу є раптові напади абдомінальноі коліки, панкреатит, гепатоспленомегалія, ксантоми на шкірі. Атеросклероз у таких осіб не розвивається. Людям з першим типом гіперліпопротеїдемій слід рекомендувати обмежене споживання жиру – до 25-35 г в день (0, 5 г на 1 кг маси тіла), з рівномірним зменшенням насичених і ненасичених жирних кислот. Вуглеводи можна споживати в звичайних кількостях. Білки обмежувати не слід. Медикаментозна корекція цього типу неефективна.
| Таблиця 2. | ||||||||
| Класифікація гіперліпопротеїдемій (на основі класифікації Д. Фредріксона) | ||||||||
| Тип | Загальний холестерин плазми крові | Холестерин ЛПНГ | Триацил-гліцерини | Аномалії ліпопротеїдів | Первинні причини (фенотип) | Вторинні причини (фенокопії) | ||
| І. Родинна гіперхіломікронемія | Підвище-ний (незначно) | Низький або в нормі | Підвищені (різко) | Надмір хіломікро-нів | Дефіцит ліпопротеїдліпази | Системний червоний вовчак, панкреатит, цукровий діабет, гіпотиреоз, прийом контрацептивів | ||
| ІІа. Родинна гіперхолестеринемія | Підвище-ний | Підвищений | Норма | Надмір ЛПНГ | Рецептори до ЛПНГ відсутні (гомозиготи) або їх кількість знижена (гетерозиготи) | Нефротичний синдром, цукровий діабет | ||
| ІІб Родинна комбінована (змішана гіперліпопротеїдемія) | Підвище-ний | Підвищений | Підвище-ний | Надмір ЛПНГ і ЛПДНГ | Дефіцит рецепторів до ЛП | Часто ІХС, інфаркт міокарда в молодому віці, мікседема, нефротичний синдром, мієломна хвороба, макроглобулінемії. | ||
| ІІІ. Родинна гіперліпопротеїдемія (дисбеталіпопротеїдемія | Дефіцит апо-Е-рецепторів | Атеросклероз судин нижніх кінцівок, ІХС, ксантоми, цукровий діабет, ліпоїдна дуга рогівки | ||||||
| ІІІ. Родинна гіпертриацилгліцери-немія Гіперпребеталіпопро-теїдемія | Нормальні | Нормальні | Підвищені | Посилене утворення і сповільне-ний кліренс ТГ, ЛПДНГ | Дефіцит апо-С. Низька активність ліпопротеїдліпази. Підвищений синтез ТГ в печінці | ІХС та інфаркт міокарда. Гіпотиреоз. Цукровий діабет. Нефротичний синдром, панкреатит. Алкоголізм. Напади абдомінальної коліки. | ||
| ІІІ. Родинна гіпертриацилгліцери-немія Гіперхіломікронемія і гіперпребеталіпопро-теїдемія | Підвище-ний | Підвищений | Дуже підвищений | Генетичний дефект кліренсу ЛП | Цукровий діабет. Ожиріння. Панкреатит Часті напади абдомінальної коліки. Алкоголізм Гепотолієнальний синдром | |||
 |
Рис. 4. Морфогенез та клінічні прояви атеросклероза
(за McGill et al., 1963).
Родинна гіперхолестеринемія – ГЛП ІІ-го типу. Це найбільш серйозна патологія обміну ліпідів, ступінь ризику розвитку ІХС в 10-20 разів вищий в порівнянні зі здоровими людьми. В плазмі крові таких людей різко підвищується вміст холестерину і ЛПНГ, особливо у гомозигот, для яких характерна повна відсутність рецепторів до апо В-ЛП (гетерозигот є лише половина нормально функціонуючих рецепторів). У гомозигот з ІІа типом гіперліпопротеїдемій рівень холестерину сягає 18, 08–25, 84 ммоль/л, а у гетерозигот 7, 75–12, 92 ммоль/л. При цьому рівень триацилгліцеринів залишається нормальним.
Клінічно родинна гіперхолестеринемія ІІа типу гіперліпопротеїдемій проявляється ознаками ІХС, інфаркта міокарда в дитячому та молодому віці.
В раціоні таких хворих має бути мінімальна кількість холестерину (менше 300 мг в день). З раціону слід вилучати продукти багаті на холестерин, обмежується споживання насичених жирів і збільшується доза ненасичених (1: 2). Кількість білків і вуглеводів звичайна. Якщо немає зайвої ваги, то калорійність іжі не слід обмежувати.
Медикаментозне лікування слід розпочинати з аніонообмінних смол (секвестрантів жовчних кислот), при їх неефективності приєднують інгібітори ГМГ-КоА-редуктази. Інгібітори ГМГ-КоА-редуктази (ловастатин, симвастатин, зокор) блокують синтез холестерину. За умов інгібування ГМГ-КоА-редуктази зростає експансія рецепторів до ЛПНГ в печінці (up-regulation) та збільшується захоплення ЛПНГ з крові. По відношенню до гетерозигот інгібітори ГМГ-КоА-редуктази є високоефективними. В той же час відносно гомозигот, у яких рецептори до ЛПНГ відсутні або ж є спадковий дефект структури апопротеїнів В-100, ефективність застосування цієі групи препаратів низька. Інгібітори ГМГ-КоА-редуктази є ефективні і у хворих з вторинними гіперліпопротеїдеміями, особливо з цукровим діабетом, хронічними захворюваннями нирок.
У хворих з гомозиготною гіперліпопротеїдемією ІІа типу кількість атерогенного холестерину ліпопротеїдів низькоі густини можна зменшити за допомогою екстракорпоральних методів: плазмаферезу, ЛПНГ-аферезу, екстракорпоральної преципітації, ЛПНГ гепарином.
Родинна комбінована (змішана) гіперліпідемія (ГЛП ІІб типу). Цей тип характеризується підвищенням рівнів холестерину сироватки крові вище 6, 2 ммоль/л та триацилгліцеринів до 2, 8-5, 6 ммоль/л. Ризик виникнення ІХС великий.
Дієтичні рекомендаціі хворим з гіперліпопротеїдеміями IIб типу подібні до таких, які даються при IIа типі гіперліпопротеїдемій. Споживання холестерину обмежують максимально, кількість жирів зменшують за рахунок насичених жирів. Кількість вуглеводів не повинна перевищувати 5 г на 1 кг маси тіла. Білки не обмежують. Алкоголь необхідно виключити повністю.
Оскільки у таких пацієнтів має місце як гіперхолестеринемія, так і гіпертриацилгліцеринемія, то медикаментозну корекцію слід проводити з використанням інгібіторів ГМГ-КоА-редуктази (інгібування синтезу ХС) і фібратів (стимуляція катаболізму ЛПДНГ шляхом активаціі ліпопротеїдліпази, інгібування утворення ЛПДНГ в печінці). Можуть бути використані і препарати риб’ячого жиру, зокрема, максепа, споживання якої у хворих з ІІб типом гіперліпопротеїдемій сприяє зниженню холестерину на 27 %, а триацилгліцеринів – на 64 %.
Родинна гіперліпопротеідемія (дисбеталіпопротеїдемія) - ГЛП ІІІ-го типу. Характерною особливістю ІІІ-го типу гіперліпопротеїдемій є високий ступінь ураження атеросклеротичним процесом всього судинного русла з клінічними проявами після 20 років. Поряд з коронарною хворобою у таких хворих є атеросклероз судин нижніх кінцівок з явищами cladicatio intermitens (минаюча кульгавість), шкірні ксантоми, ксантелазми, в області ліктів, колін, сідниць, ліпоїдна дуга рогівки. У таких хворих порушена толерантність до глюкози, навантаження вуглеводами може викликати різку гіперліпідемію; у них часто є цукровій діабет.
Приступаючи до лікування, хворий насамперед мусить подбати про зайву вагу, бо часто нормалізація маси тіла вже сама по собі сприяє зниженню рівня холестерину і триацилгліцеринів плазми крові. Кількість вуглеводів не повинна перевищувати 4 г на кг маси тіла. Білки не обмежують. Алкоголь виключається. Медикаментозне лікування починають з нікотиновоі кислоти, при значній гіперхолестеринемії застосовують інгібітори ГМГ-КоА-редуктази (симвастатин, зокор).
Родинна гіпертриацилгліцеринемія – ГЛП ІV типу, - відрізняється від І типу (гіперхіломікронемії) тим, що підвищення рівня триацилгліцеринів в плазмі крові відбувається за рахунок фракції ЛПДНГ (ендогенна триацилгліцеринемія), акумуляції хіломіронів при цьому не спостерігається. Зустрічається лише у дорослих.
Клінічні прояви родинної гіпертриацилгліцеринемії не є строго специфічними; можуть спостерігатись ознаки ураження як вінцевих, так і периферійних судин. Атеросклероз розвивається повільно. Іноді спостерігаються напади абдомінальної коліки або панкреатиту. Фенокопії цього типу спостерігаються при надмірному споживанні алкоголю, нелікованому цукровому діабеті, гіпотиреозі, нефротичному синдромі, панкреатиті, диспротеїнеміях, ожирінні.
Дієтичні рекомендації: насамперед виключити споживання алкоголю і нормалізувати масу тіла. В разі нормальної маси тіла жир не обмежувати, віддаючи однак перевагу ненасиченим жирам. Кількість холестерину не повинна перевищувати 300-500 мг на добу, а вуглеводів – 4 г на 1 кг маси тіла. Білки не обмежуються.
Медикаментозна корекція. Препаратами вибору є фібрати (уфібрат, гемфіброзил), препарати жиру оселедця (максепа), омега-3-жирних кислот (ейкозопентаєнової та докозагексаєнової) та нікотинова кислота.
Родинна гіпертриацилгліцеринемія – ГЛП V типу. При цьому поряд з гіпертриацилгліцеринемією має місце гіперхіломікронемія. У цих хворих є генетичний дефект кліренсу ліпопротеїнів, багатих на триацилгліцерини.
Ризик ішемічної хвороби серця у них лише дещо підвищений, головна ж мета лікування – попередження панкреатиту.
У таких хворих, як правило, збільшена печінка і селезінка, часті напади абдомінальної коліки (особливо після переїдання жирами), часті загострення хронічного панкреатиту.
При цьому типі гіперліпопротеїдемій обмеження жиру в раціоні менш строге, як при І типі; 30% калорійності їжі може припадати на жири. Проте, якщо у хворого є приступи панкреатиту чи абдомінальної коліки, то доля жиру в раціоні мусить бути ще меншою (15-20%). Жири повинні бути рослинні і складатися, в основному, з поліненасичених жирних кислот. Кількість холестерину обмежують до 300-500 мг/добу, вуглеводів – 5 г/кг маси тіла при нормальній вазі, в разі ж зайвої ваги – кількість вуглеводів зменшують до 4 г/кг маси тіла. Алкоголь не рекомендується.
З медикаментозних середників препаратами вибору є нікотинова кислота і фібрати (уфібрат, геміфіброзил).
Клініка і діагностика. Клінічні ознаки атеросклерозу зумовлені ступенем звуження просвіту артерії. Ранні атеросклеротичні зміни в судинах не супроводжуються клінічними симптомами, тому зазвичай не діагностуються.
Початкові ознаки атеросклерозу є загальними: схильність до регіонарних спазмів, збільшення кількості холестерину і/або триацилгліцеринів у крові тощо.
Клінічні ознаки атеросклерозу за О.Л. Мясниковим є наступними:
I. Доклінічний, скритий період, в якому спостерігаються вазомоторні метаболічні зміни;
II. З клінічними проявами: перша стадія – ішемічна (звуження судин, яке призводить до порушення харчування і дистрофічних змін у відповідних органах); друга стадія – тромбонекротична: некрози, дрібновогнищеві або великі (з тромбозом судин або без них); третя стадія – склеротична або фіброзна: розвиток фірозних (шрами) змін в органах з атрофією їх паренхіми.
Великого значення О.Л.Мясников надає доклінічному періоду, який перебігає з нервово-судинними порушеннями й схильністю до загальних і регіонарних спазмів або тонічним скороченням судин, а також змінами у складі ліпідів і ліпопротеїнів із збільшенням фракції b-ліпопротеїнів.
Щодо загальних ознак атеросклерозу, то насамперед звертає на себе увагу передчасне постаріння хворого на атеросклероз. Шкіра стає сухою, витонченою, з пониженим тургором, зморщеною. Артерії стають звивистими, їх добре стає видно на згинальній частині ліктів, внутрішніх поверхнях плечей, на скронях. Внаслідок втрати еластичності їх стінки при пальпації нагадують “гусяче горло”, вони нерівномірно ущільнені, складають враження вервиці. Знижується швидкість проходження хвилі крові крізь ущільнені артерії. Дрібні вени, зазвичай, розширюються. Часто знаходять різне наповнення пульсу на лівих і правих aa. radiales et dorsales pedis.
У випадках атеросклерозу судин головного мозку проводять транскраніальну доплерографію з дослідженням сонних (загальних, зовнішніх та внутрішніх), вертебральних, передньо-, середньо- та задньомозкових, надблокових артерій. З допомогою доплерографії добре діагностується також атеросклеротичне пошкодження коронарних, ниркових та магістральних судин кінцівок.
Атеросклероз грудної аорти. Типових клінічних ознак при цьому нема, проте з розвитком недостатності кровоплину з’являються задишка і серцебиття. При атеросклерозі коронарних артерій виникає загрудинний біль (ІХС). Разом з тим, загрудинний біль з поширенням в ліву руку (рідше праву) та на ділянку шиї може спостерігатись при аортоалгії внаслідок подразнення plexus aorticus. Біль при аорталгії, проте, немає чіткого вираженого приступоподібного характеру, є тривалим, посилюється при фізичному навантаженні. Характерним для атерослерозу аорти є підвищення систолічного і зниження діастолічного артерільного тиску. Частою ознакою є симптом ретростернальної пульсації, яка зумовлена інтенсивною пульсацією розширеної і видовженої аорти. У багатьох хворих при цьому знаходять посилену пульсацію підключичної артерії – симптом Трунечека. Перкуторно виявляють ознаки розширення аорти – зона притуплення виступає на 2-3 см вправо від краю грудини.
При аускультації вислуховується систолічний шум над аортою. “Склеротичний” шум зазвичай є грубим, він посилюється після фізичного навантаження, часто краще вислуховується на грудині в напрямку до яремної ямки. ІІ тон над аортою дзвінкий (“акцентований”), часто є з металічним відтінком, до того ж при значному розширенні аорти він сильно резонує. Систолічний шум зумовлений нерівністю внутрішньої поверхні стінки аорти внаслідок ураження її атеросклеротичним процесом (бляшками), а також атеросклеротичним потовщенням стулок аортального клапана. Систолічний шум зазвичай посилюється, якщо запропонувати хворому покласти руки за голову і нагнути голову трохи допереду – симптом Сиротиніна-Куковерова. При рентгенологічному дослідженні виявляють значне потовщення аорти, її дуги, посилення інтенсивності тіні або ж дифузне розширення аорти та її розгорненість. У осіб старше 50 років при рентгенологічному дослідженні за атеросклероз аорти може бути прийнята аорта з кальцинозом її середньої та внутрішньої оболонок (неатерогенний кальциноз аорти – форма Менкеберга), але у таких випадках збільшення поперечника і видовження аорти зазвичай невелике. При кальцинозі аорти тінь судини більш інтенсивно із щільним обрамленням за контуром аорти.
Атеросклероз аорти може тривалий час не спричиняти порушення кровоплину. Зазвичай недостатність кровоплину з’являється тоді, коли до аортосклерозу долучається кардіосклероз (дифузний).
Атеросклероз черевного відділу аорти є частою локалізацією атеросклерозу. При цьому є нерівномірне потовщення стінки аорти, що складає враження її звивистого характеру при пальпації. Остання можлива лише при тонкій податливій кволій передній черевніій стінці. З допомогою рентгенологічного дослідження знаходять вогнища кальцинозу черевної аорти. Діагноз підтверджує ультразвукове дослідження черевної аорти.
Атеросклероз легеневої артерії. Зазвичай розвивається вторинно при хворобах серця і легень, при яких упродовж багатьох років підвищений кров’яний тиск в легеневій артерії (стеноз лівого венозного отвору, відкрита аортальна протока, хронічні обструктивні хвороби легень тощо).
Атеросклероз ниркових артерій зустрічається порівняно часто і призводить до ішемії ниркової ттканини та підвищення тиску у великому колі кровоплину – реноваскулярної гіпертензії. Атеросклеротичні бляшки можуть локалізуватись не лише у головних ниркових артеріях, котрі відходять від черевного відділу аорти, але й у їх розгалуженнях - аж до аа. іnterlobularis et arcuatae. Патологічні зміни в нирках внаслідок атеросклеротичного стенозу артерій нирок носять зазвичай вогнищевий характер і не призводять до виражених порушень функцій нирок. На аутопсії нирка – з великими горбами, зменшена в об’ємі.
При житті у таких хворих, окрім гіпертензії, виявляють зміни в сечі – невелику кількість білка, еритроцитів, циліндрів. Атеросклероз нирок слід відрізняти від артеріолосклерозу нирок, який є фінальною стадією гіпертензивної хвороби. У таких випадках знаходять гіаліноз в а. іnterlobularis, v.afferentia, капілярах клубочків. Артеріолосклероз нирки – це не вогнищеве, а дифузне ураження нирок, яке носить назву “первинно зморщеної нирки”. Вона викликає стійку ренопаренхіматозну і/або ренопривну артеріальну гіпертензію, яка супроводжується прогресуючою хронічною нирковою недостатністю. “Первинно зморщену нирку” необхідно диференціювати із “вторинно зморщеною ниркою”, яка є наслідком гломеруло- або пієлонефриту.
Атеросклероз мезентеріальних артерій. Зазвичай атеросклеротичний процес локалізуєтьсяу верхніх брижових артеріях. При цьому хворого турбують напади інтенсивного болю у надчерев’ї та мезогастрії, які нагадують загрудинний больовий синдром. Такий симптомокомплекс носить назву “angina abdominalis”. Тепер у таких випадках ставлять діагноз “ішемічна хвороба черевної порожнини” (по аналогії з терміном “ішемічна хвороба серця”). Стенокардитичний біль у животі супроводжується вираженим метеоризмом, закрепами (обстипаційний синдром). Він є результатом склерозування а.mesenterica superior. У випадках дестабілізації мезентерального кровоплину розвивається клініка синдрому “гострого живота”, який вимагає хірургічного лікування.
Атеросклероз артерій підшлункової залози. Є частою локалізацією ішемічної хвороби черевної порожнини При цьому зазвичай порушується харчування острівців Лангерганса, наслідком чого є розвиток цукрового діабету, глюкозурії. У таких хворих є підвищений апетит, швидко розвивається абдомінальний тип ожиріння.
Атеросклероз артерій нижніх кінцівок. Викликає недостатнє кровопостачання м’язів, що проявляється типовим синдромом “минаючої кульгавості” (claudicatio intermittens), спастичне скорочення м’язів кінцівки (або й обох кінцівок). В час ходи з’являється відчуття “оніміння” ніг, “повзання мурашок”, біль в ногах, яка змушує хворого зупинитись. Після відпочинку порушений кровоплин відновлюється і хворий йде дальше, але біль знову відновлюється і він знову зупиняється тощо. У окремих хворих після декількох зупинок подальша хода вже не викликає болю, і пацієнт помалу долає певну відстань (феномен “розходжування”). При пальпації склерозованої судини часом знаходять швидкий підйом пульсової хвилі, що добре видно на сфігмограмі або п’єзограмі артеріального пульсу. Швидкий підйом пульсової хвилі пояснюється ригідністю стінки артерій. Швидкість поширення пульсової хвилі є тим більшою, чим виражене атеросклеротичне ураження стінки артерій. Атеросклероз великих артеріальних стовбурів добре діагностується з допомогою сонографії та доплерографії.
Лікування. Загальновідомо, що лікування хворого з проявами (біохімічними або клінічними чи клініко-біохімічними) атеросклерозу треба розпочинати з досить агресивних дієтичних рекомендацій. Метою дієти є зниження рівня атерогенних ліпопротеїдів нижче критичних показників, при яких слід призначати медикаментозні засоби.
Експерти Європейського товариства боротьби з атеросклерозом рекомендують наступні заходи: 1) зменшити на 10% загальне вживання жирів; 2) різко зменшити вживання насичених жирних кислот (тваринні жири, масло, вершки, сир, яйця, м’ясо); 3) збільшити вживання продуктів, багатих поліненасиченими жирними кислотами (рідкі рослинні олії, риба, птиця, морські продукти); 4) збільшити вживання клітковини та складних вуглеводів (овочі, фрукти, крупи); 5) замінити при домашньому готуванні страв масло, маргарин рослинними оліями (рослинні жири показані при всіх типах гіперліпопротеїдемій, за винятком І, при якому їх обмін порушений так само, як і насичених жирних кислот); 6) різко зменшити вживання продуктів, багатих на холестерин; 7) різко зменшити кількість кухонної солі в їжі, що споживається.
Медикаментозне лікування атеросклерозу. Медикаментозне лікування атеросклерозу слід застосовувати лише у випадку, коли дієтичні заходи протягом 3-6 місяців виявились не ефективними. Дотримуватись дієти необхідно також підчас прийому гіполіпідемічних препаратів.
Метою дієтотерапії та медикаментозного лікування є зниження загального холестерину до 2, 9-3, 0 ммоль/л. При такій концентрації зберігається нульовий баланс холестерину в судинній стінці; при більш високій концентрації він відкладається в інтимі артерій.
В разі гіпертриацилгліцеринемії метою лікування є попередження синдрому хіломікронемії з явищами панкреатиту або важкого абдомінального больового синдрому.
З допомогою гіпохолестеринемічної терапії можна домогтися: 1) профілактики атеросклерозу, якщо лікування проводиться в дитячому віці; 2) затримки подальшого розвитку атеросклеротичних уражень, якщо лікування розпочато в дорослому віці; 3) видалення холестерину з бляшок, що робить їх менш небезпечними в плані утворення на їх поверхні тромбів; 4) частковій або значній регресії атеросклерозу.
Приступаючи до медикаментозного лікування атеросклерозу необхідно з’ясувати, який тип гіперліпопротеїдемії має місце у хворого.
Всі гіполіпідемічні засоби, які сьогодні використовуються, поділяються на дві групи:
І. Препарати з переважаючим впливом на загальний холестерин плазми і холестерин ліпопротеїдів низької густини – інгібітори ГМГ-КоА-редуктази; секвестранти жовчних кислот; пробукол.
ІІ. Препарати з переважаючим впливом на рівень плазменних триацилагліцеринів і триацилагліцеринів ліпопротеїдів дуже низької густини (фібрати; нікотинова кислота; поліненасичені омега-3-жирні кислоти – ейкозопентаєнова і докозогексаєнова; максепа).
Гіполіпідемічні препарати повинні прийматись пацієнтами впродовж всього життя з моменту виявлення порушення ліпідного обміну. Вкрай важливо, щоб гіполіпідемічні засоби, знижуючи рівень ЛПНГ і ЛПДНГ у крові, не знижували б, а, ще краще, підвищували рівень антиатерогенних ЛПВГ.
Інгібітори ГМГ-КоА-редуктази (3-гідрокси-3-метил-глютарил-коензим-А-редуктази). ГМГ-КоА-редуктаза – це ключовий фермент-каталізатор біосинтезу холестерину. В нормі з ГМГ-коензиму А утворюється мевалонова кислота та ізопреноїди, які використовуються для синтезу холестерину. Деякі види грибів (Aspergillius terreus) синтезують речовини, які здатні блокувати синтез холестерину на стадії перетворення ГМГ-КоА в мевалонову кислоту. Такі речовини отримали назву статинів. В клінічній практиці тепер застосовується декілька препаратів групи статинів: ловастатин (мевакор), симвастатин (зокор), правастатин (правакол), флувастатин (лескол).
За хімічною структурою статини подібні до ГМГ-КоА-редуктази, вони пригнічують цей фермент за конкурентним принципом. Статини, вжиті всередину, нагромаджуються в печінці, де й гальмують синтез холестерину. Це приводить до зниження вмісту холестерину в клітинах печінки і утворення додаткової кількості рецепторів на плазматичних мембранах до ЛПНГ (апо В, Е-рецепторів). Як наслідок, збільшується захоплення гепатоцитами ЛПНГ для забезпечення клітин печінки холестерином, необхідним для утворення жовчних кислот. Це призводить до зниження рівня ЛПНГ і холестерину в крові. Важливо, що статини не повністю блокують активність ГМГ-КоА-редуктази, тому в клітині зберігається біологічно необхідний рівень мевалонової кислоти. Не порушують статини й функцію статевих залоз та наднирників.
Найбільш поширеним серед статинів є сьогодні ловастатин та симвастатин. Початкова доза симвастатину 10 мг/день, одноразово ввечері. При легкій або помірній гіперхолестеринемії початкова доза може складати 5 мг 1 р/день. Через 4 тижні добова доза, в разі необхідності (коли рівень холестерину > 5, 2 ммоль/л), збільшують до 20 мг/добу. Рідко виникає необхідність у збільшенні добової дози до 40 мг. Екскретується симвастатин з жовчю. В ході багатоцентрового Скандинавського рандомізованого плацебо-контрольованого дослідження впливу симвастатину на виживання (Scandinavian Simvastatin Survival Study, 4S) знайдено, що зокор знижує ризик смертності на 30%.
Протипоказами до застосування статинів є ідіосинкразія, захворювання печінки в активній фазі, вагітність, період лактації.
Секвестранти жовчних кислот (аніонообмінні смоли). Це синтетичні аніонообмінні смоли, які не всмоктуються в шлунково-кишковому тракті. Найбільш поширені 2 представники цього класу – холестирамін та колестипол, обидва є кополімерами. Препарати аніонообмінних смол зв’язуються в просвіті кишечника з жовчними кислотами, обриваючи в такий спосіб їх кишково-печінкову циркуляцію (в нормі 95% жовчних кислот реабсорбується). Холестирамін здатний збільшувати виділення жовчних кислот з калом в 30 разів. За таких умов, печінка змушена посилювати синтез жовчних кислот. Для цього їй необхідний холестерин, тому-то гепатоцити посилюють вилучення ЛПНГ з крові. З цією метою збільшується синтез рецепторів до ЛПНГ (апо В-, Е- рецепторів). Як наслідок, посилюється катаболізм ЛПНГ рецепторним шляхом, рівень загального холестерину і холестерину ЛПНГ знижуться на 15-20%. Небажаним ефектом аніонообмінних смол є підвищення в плазмі рівня триацилгліцеринів.
Аніонообмінні смоли, подібно до статинів, ефективні при гетерозиготній родинній гіперхолестеринемії. Позитивні вони також при неспадковій гіперхолестеринемії ІІа типу ГЛП. Проте вони цілком неефективні при гомозиготній гіперхолестеринемії, коли клітини не мають рецепторів до ЛПНГ взагалі. Аніонообмінні смоли в якості монотерапії не показані в разі гіпертриацилгліцеринемії, тобто при І-ІV і V типах ГЛП. Для усунення небажаного підвищення триацилгліцеринів їх краще поєднувати із статинами або з нікотиновою кислотою чи фібратами. Аніонообмінні смоли призначають у великих дозах: холестирамін 16-24 г/добу, колестипол до 10 г/добу. Добову дозу слід поділити на 2-4 порції і приймати з їжею або фруктовим соком. Препарат вазозан П – це холестирамін з поліпшеними смаковими якостями.
Небажані ефекти: закрепи, нудота, відчуття дискомфорту в черевній порожнині, порушення всмоктування фолієвої кислоти і жиророзчинних вітамінів (тому їх слід приймати одночасно із статинами).
Пробукол. Це розчинна в жирах сполука, всмоктується з кишково-шлункового тракту не повністю (10%), краще всмоктується при споживанні жирної їжі. Після всмоктування зв’язується з хіломікронами та ЛПДНГ, нагромаджується у жировій тканині. Виводиться з організму повільно – до 6 міс. після завершення курсу лікування. Відомо, що ЛПНГ атерогенними стають тоді, коли піддаються модифікації, зокрема шляхом перекисного окислення. Пробукол блокує оксидацію ЛПНГ, прискорює катаболізм холестерину. Призначається в дозі 500 мг 2 рази денно разом з їжею. Лікування проводиться курсами по 3-6 міс. з 6-місячними перервами.
“Есенціальні” фосфоліпіди – це високоочищений екстракт з бобів сої, особливістю їх є наявність в них великої кількості поліненасичених жирних кислот, особливо ліноленової (70%). Завдяки особливостям хімічної структури ліпостабіл (есенціале) вбудовується в мембрани клітин і клітинних органел, зокрема, в поверхневий пласт ліпопротеїдних часток ЛПВГ, знижуючи їх мікров’язкість та підвищуючи фізіологічну активність. Вбудовуючись в мембрани еритроцитів, ліпостабіл (есенціале) понижує в’язкість крові, а в тромбоцити – їх агрегаційні властивості. Есенціальні фосфолпіди адсорбують з мембран холестерин, тобто виконують функцію ЛПВГ, внаслідок чого посилюється “зворотний транспорт холестерину”.
В одній ампулі (10 мл) ліпостабілу є: есенціальних фосфоліпідів (природнього походження з переважанням ненасичених жирних кислот, особливо лінолевої (70%), ліноленової та олеїнової кислоти) - 500 мг, вітаміну В6 – 4 мг, нікотинової кислоти – 2 мг, АМФ – 2 мг. В одній ампулі (5 мл) есенціале є: “есенціальних” фосфоліпідів – 250 мг, пірідоксин хлориду – 2, 5 мг, ціанокобаламіну – 10 мг, натрію д-антотенату – 1, 5 мг, нікотинаміду – 25 мг. Вводяться ці препарати довенно краплинно в 250 мл 5% розчину глюкози або струменево без розведення, “на крові”. Курс довенних ін’єкцій 20-30 днів, в подальшому хворого слід перевести на пероральний прийом ліпостабілу (есенціале) по 1-2 капсулі 3-4 рази на день впродовж 2-3 міс.
Фібрати. Це деривати ізомасляної кислоти, з яких практичне значення мають клофібрат, фенофібрат (ліпантин), гемфіброзил, уфібрат. Фібрати підвищують активність ліпопротеїнліпази і тим самим прискорюють катаболізм ЛПДНГ, посилюють синтез рецепторів до ЛПНГ, інгібують утворення ЛПДНГ у печінці. При прийомі фібратів рівень триацилгліцеринів може знизитись навіть наполовину, при цьому рівень ЛПВГ зростає на 25%. Фібрати показані при змішаних гіперліпопротеїдемііях, зокрема при ІІб-ІV і V типах. При ІІІ типі ГЛП, вони є препаратами вибору. Гемфіброзил більш ефективно знижує рівень тригліцеридів при ІV типі ГЛП. Фібрати застосовують в добових дозах від 300-400 мг до 2 г. Звичайно вони добре переносяться хворими, але іноді можуть викликати розлади шлунково-кишкового тракту, міозит, гепатит, холелітіаз. Найбільш часто формуються каміння в жовчному міхурі при застосуванні клофібрату. Останнє, а також збільшенння числа численних наслідків від не серцево-судинних захворювань, призвело останнім часом до значного обмеження клофібрату.
Більш безпечним є гемфіброзил (добова доза 900 мг) та фенофібрат або ліпантин (900-1200 мг/добу), які сьогодні рекомендуються в разі переважання гіпертриацилгліцернемії над гіперхолестеринемією.
Нікотинова кислота. Як і фібрати володіє здатністю знижувати в крові передусім рівень триацилгліцеринів. Під впливом нікотинової кислоти у великих дозах (2, 4-4 г/день) рівень триацилгліцернемії знижується на 20-60%, менш виражене при цьому зниження концентрації холестерину. Важливо, що одночасно зростає в крові рівень антиатерогенних ЛПВГ. Нікотинова кислота найбільш ефективна при ІІІ і V типах ГЛП; при ІІб і IV ефект її менш виражений. З небажаних ефектів, які обмежують застосування нікотинової кислоти необхідно відмітити різку гіперемію шкіри (посилення притоку крові до голови, обличчя), відчуття жару, свербіння шкіри, кропив’янка, пониження толерантності до вуглеводів, почастішання приступів стенокардії (виникає внаслідок тахікардії і підвищення споживання міокардом кисню). Для попередження небажаних ефектів лікування нікотиновою кислотою починають з малих доз (100-200 мг 3-4 рази на день), лише згодом (через 2 тижні) переходять поступово до максимальних доз, які й дають гіполіпідемічний ефект.
Є декілька похідних нікотинової кислоти, в яких небажані ефекти зустрічаються рідше і слабше виражені. Це препарати аципимокс (750-1500 мг/добу, в 3-4 прийоми) і ендурацин – таблетки пролонгованої дії.
Препарати риб’ячого жиру. В останні роки почали широко використовуватись препарати, які містять в собі омега-3 ейкозопентаєнову і омега-3 докозогексаєнову кислоти. Вони приймаються в добовій дозі 2-3 г, що призводить до зниження утворення ЛПДНГ в печінці і до зниження рівня цього класу ліпопротеїдів у пацієнтів з IV і V типами ГЛП. Вказані кислоти гальмують агрегацію тромбоцитів, тобто поліпшують реологічні властивості крові. Препарат з риб’ячого жиру максепа зарекомендував себе як ефективний середник при гіпертриацилгліцернемії в поєднанні з гіперхолестеринемією (ІІб тип ГЛП).
Немедикаментозні методи корекції гіперхлестеринемії. Екстракорпоральне вилучення етерогенних апоВ-вмісних ліпопротеїнів. Це досягається плазмаферезом або селективною сорбцією ЛПНГ-“аферез ЛПНГ”. До цих методів вдаються в разі неефективності дієтотерапії та медикаментозного лікування. Частіше такі методи використовуються у випадках гомозиготної гіперхолестеринемії. Аферез ЛПНГ характеризується великою вибірковістю, адже в процесі цієї процедури видаляються тільки апо-В-ЛП, головним чином ЛПНГ. Селективне видалення ЛПНГ здійснюється за допомогою моноклональних антитіл до ЛПНГ (точніше до апо-В). За одну процедуру можна вивести з організму понад 10 г холестерину. Процедури аферезу ЛПНГ необхідно періодично повторювати. Їх треба поєднувати з прийомом гіполіпідемічних препаратів. При такому лікуванні через 1, 5-2 роки вдається досягти часткової регресії атеросклеротичних уражень вінцевих артерій.
Генно-інженерні підходи до лікування атеросклерозу. Суть генетичної корекції гіперліпопротеїдемій полягає в переносі генів, які контролюють синтез тих чи інших апопротеїнів або апо-В, Е-рецепторів. Перші спроби генетичної корекції (генотерапії) спадкового дефекту апо-В, Е-рецепторів вже здійснені у США. Принцип генотерапії полягає у наступному. Проводять резекцію 15% маси печінки пацієнта, видалені з біоптата печінкові клітини вирощують в умовах культивування, потім з допомогою рекомбінантних ретровірусів в ці клітини переносять функціонуючий ген апо-В, Е-рецептора. Відтак гепатоцити з імплантованими в них генами повертають хворому шляхом ін’єкції в ворітну вену. Регенерація видаленої частини печінки сприяє експресії генів апо-В, Е-рецепторів. На думку американськиих вченів цей метод є досить перспективним.
Таким чином, викладені в цьому розділі матеріали щодо етіології, патоморфології, патогенезу, клінічних атеросклерозу та можливостей сучасної антисклеротичної терапії не залишають сумніву в тому, що вже в недалекому майбутньому медицина буде спроможна повністю контролювати атерогенез на всіх етапах його становлення.
|
|