Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Лекция 5
|
|
Кинетика гетерогенных химических реакций. Теория активных (бимолекулярных) столкновений. Теория активного комплекса. Катализ: основные положения, гомогенный и гетерогенный катализ, активные центры на поверхности катализатора.
5.1. КИНЕТИКА ГЕТЕРОГЕННЫХ ХИМИЧЕСКИХ РЕАКЦИЙ.
Гетерогенные реакции – это химические реакции, протекающие на границе раздела фаз (чаще всего раствор/твердая фаза или газ/твердая фаза) в тонком диффузном слое. Если пренебречь процессами сорбции и десорбции веществ на границе раздела фаз, то можно выделить три стадии подобных процессов:
1) подвод исходных веществ из объема раствора к поверхности раздела фаз;
2) протекание химической реакции в тонком диффузном слое;
3) отвод продуктов реакции вглубь раствора.
Ограничимся рассмотрением случая, когда перенос вещества осуществляется только за счет диффузии — переноса вещества, обусловленного неравенством химических потенциалов внутри системы (вызванного разностью концентраций веществ в системе). В процессе диффузии вещество переходит из области с большей концентрацией в область с меньшей концентрацией (процесс направлен в сторону уменьшения химического потенциала вещества). Процессы диффузии в заданном направлении с кинетической точки зрения описываются законом Фика: количество вещества dn, проходящее за время dt через площадь поверхности S, прямо пропорционально градиенту концентрации вещества dC/dx в растворе:
 , (5.1)
, (5.1)
или
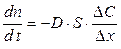 , (5.2)
, (5.2)
где Δ С — разность концентраций вещества в точках, отстоящих друг от друга на расстояние Δ х, т. е. на толщину диффузионного слоя. Коэффициент пропорциональности D называют коэффициентом диффузии, он равен количеству вещества, прошедшего за единицу времени через единицу площади поверхности при градиенте концентрации, равном единице. Коэффициент диффузии увеличивается с повышением температуры по закону, аналогичному уравнению Аррениуса:
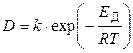 ,
,
где Е д — энергия активации процесса диффузии, редко превышающая величину в 4 ‑ 10 кДж/моль, т. е. во много раз меньше энергий активации большинства химических реакций.
Рассмотрим случай, когда объем раствора не меняется в ходе химической реакции (V = const), химическая реакция протекает на поверхности раздела фаз; концентрация вещества меняется в диффузионном слое от значения С (концентрации вещества в объеме раствора) до значения СS (концентрации вещества в приповерхностном слое). Скорость растворения твердых тел определяется скоростью диффузии растворяемого вещества из поверхностного слоя в жидкую фазу
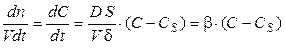 , (5.3)
, (5.3)
где β = (D · S)/(V δ) — константа скорости диффузии или константа массопередачи, δ – толщина диффузионного слоя. Скорость процесса диффузии прямо пропорциональна разности концентраций вещества в объеме раствора и приповерхностном слое.
Рассмотрим мономолекулярную реакцию А → В, протекающую в гетерогенных условиях. С течением времени в системе при определенных давлении и температуре устанавливается стационарное (не изменяющееся во времени) состояние, при котором скорости собственно химической реакции (w 1), диффузии исходного вещества из объема раствора в поверхностный слой (w 2) и химической реакции в целом (т. е. скорости образования вещества В — w) становятся равными:
w = 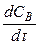 = w 1 = w 2.
= w 1 = w 2.
Скорость химической реакции w 2 зависит от концентрации вещества А на поверхности раздела фаз (CS):
w 2 = k·CS. (5.4)
В стационарных условиях скорость процесса диффузии и скорость собственно химической реакции равны:
w 1 = b · (C – CS) = w 2 = k·CS.
Тогда
 . (5.5)
. (5.5)
Подставив выражение (11.5) в ур-е (11.4), получим
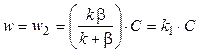 . (5.6)
. (5.6)
В стационарных условиях макроскопическая скорость реакции (константа k 1) соответствует уравнению реакции первого порядка по концентрации реагирующего вещества. Это уравнение было впервые получено А.Н. Щукаревым и может быть применимо для процессов кристаллизации солей из пересыщенных растворов, процессов испарения жидкостей в воздух в замкнутом сосуде и т. д.
Пример 5.1. Определить скорость растворения куска мрамора в 1, 0 н. растворе соляной кислоты:
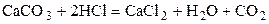 ,
,
если концентрация кислоты через один час уменьшается в 4 раза.
Решение. Если кислота поступает к твердому телу за счет диффузии, то ее концентрация вблизи твердой поверхности из-за быстрой реакции взаимодействия с мрамором падает практически до нуля. Поэтому скорость растворения мрамора определяется концентрацией кислоты в объеме (С):
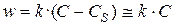 ,
,
Растворение твердых веществ идет как односторонняя реакция первого порядка. Константу скорости определим по уравнению
 мин-1,
мин-1,
скорость растворения мрамора через 1 час равна
 моль экв/(л·мин).
моль экв/(л·мин).
Гетерогенные процессы протекают в несколько стадий, скорость процесса в целом определяется скоростью наиболее медленной стадии. Поэтому выделяют три области протекания гетерогенных процессов (рис. 8):
1). Кинетическая область — независимо определенная скорость диффузии значительно превышает скорость собственно химической реакции. В этих условиях k < < β, следовательно (из ур-я 11.6) k 1 ≈ k, т. е. в стационарных условиях скорость всей реакции определя-
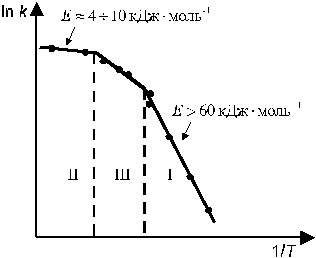 |
Рис. 8. Области протекания гетерогенных химических процессов.
I — кинетическая, II — диффузионная, III — переходная области.
ется скоростью собственно химической реакции. Если определить значение энергии активации реакции в этой области на основе температурной зависимости опытной константы скорости (k 1), то ее значение будет составлять более 60 кДж/моль (что характерно для химических реакций). В кинетической области концентрации реагирующего вещества в объеме и на поверхности раздела фаз мало отличаются и практически постоянны, т. е. СS ≈ C ≈ const.
2. Диффузионная область — скорость собственно химической реакции значительно превышает скорость диффузии, т. е. все подведенное к поверхности раздела фаз вещество А практически сразу превращается в продукт реакции. При этом концентрация А на поверхности раздела фаз очень мала (CS ≈ 0). В этих условиях k > > β, следовательно (из ур-я 11.6) k 1 ≈ β. Лимитирующей стадией реакции является диффузия реагирующего вещества к поверхности раздела фаз. Опытная энергия активации в этой области составляет менее 10 кДж/моль, что соответствует энергии активации процесса диффузии.
3. Переходная область — скорости диффузии и собственно химической реакции соразмерны между собой. В этих условиях k ≈ β, опытная энергия активации более 10 кДж/моль и увеличивается при понижении температуры.
Опытная константа скорости всей гетерогенной реакции определяется по скорости образования продукта реакции В. Следует понимать, что при стационарном протекании гетерогенной реакции термин “диффузионная область” не означает, что действительная скорость реакции на реакционной поверхности больше скорости диффузии к этой поверхности. Обе эти скорости равны, но благодаря медленному притоку веществ к поверхности скорость химической реакции на поверхности принудительно становится равной скорости диффузии.
5.2. ТЕОРИЯ АКТИВНЫХ (БИМОЛЕКУЛЯРНЫХ) СТОЛКНОВЕНИЙ.
В уравнении Аррениуса в интегральной форме
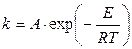 (5.7)
(5.7)
фигурируют два параметра, определяемые экспериментально на основе температурной зависимости опытной константы скорости реакции — энергия активации Е и предэкспоненциальный множитель А. Одной из задач кинетики является теоретический расчет скорости данной реакции в заданных условиях (без проведения соответствующих экспериментов), зная только концентрации и свойства исходных веществ (и продуктов реакции). Для этого необходимо уметь рассчитывать константу скорости реакции при заданной температуре, следовательно, уметь вычислять значения А и Е.
Теория активных столкновений молекул позволяет рассчитывать значения предэкспоненциального множителя А, а теория активного комплекса (или теория переходного состояния) — и значения Е, т. е. позволяет вычислять значение константы скорости при данной температуре.
Основные положения теории активных (молекулярных) столкновений:
1. Любая химическая реакция протекает через промежуточную стадию образования активных молекул реагирующих веществ (только из активных молекул могут образоваться продукты реакции).
2. Активные молекулы всегда находятся в равновесии с неактивными (нормальными) молекулами.
3. Реакция образования активных молекул эндотермична и идет с поглощением теплоты E ΄ в расчете на 1 моль активных молекул. Схематично первые три условия можно записать так
А норм. 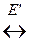 А *акт.
А *акт. 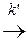 продукты
продукты
На первой стадии устанавливается равновесие меду активными и нормальными молекулами реагирующих веществ, на второй стадии из активных молекул образуются продукты реакции (константа скорости k ΄).
4. Константа скорости k ΄ не зависит от температуры, т. е. скорость реакции при повышении температуры увеличивается только за счет увеличения концентрации активных молекул.
5. Активные молекулы отличаются от неактивных только тем, что имеют некоторую избыточную энергию по сравнению со средней энергией молекул. Молекулы приобретают избыточную энергию путем соударения молекул и взаимного перераспределения энергии при этом.
Химическая реакция начинается со столкновения двух частиц. Однако, как показывают опыты, число прореагировавших частиц (молекул, ионов) при не очень высоких температурах на много порядков ниже числа сталкивающихся частиц. Так, например, в реакции:
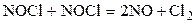
при 529 К и 1 атм образуется примерно 2·10-6 моль NO в 1 см3 за 1 с. Расчет полного числа столкновений при этих условиях приводит к значениям порядка 105 молей NO/(см3·с). Если обозначить через z число общих столкновений, а символом za – число активных столкновений, приводящих к реакции, то для указанной реакции
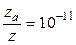 ,
,
т. е. активным столкновением будет только одно из 1011 общих столкновений.
Именно расхождения между зачениями z и za привели Аррениуса к идее о том, что в реакцию могут вступать только молекулы, обладающие определенным избытком энергии (энергией активации). Теория активных столкновений приводит к следующему соотношению между za и z:
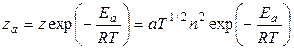 (5.8),
(5.8),
в котором учтена зависимость z от температуры Т (z = аT 1/2 n 2). В ур-и (11.8) n – число молекул в см3; Ea – так называемая “ классическая” энергия активации, равная разности между потенциальными энергиями активных и исходных молекул; а – константа, зависящая от размеров вступающих в реакцию молекул.
Сопоставляя ур-я (11.7) и (11.8) и с учетом того, что
 ,
,
получаем
 . (5.9)
. (5.9)
После логарифмирования и дифференцирования по температуре ур-я (11.9):
 , (5.10)
, (5.10)
откуда получим связь “ классической” и опытной (аррениусовской) энергией активации:
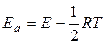 . (5.11)
. (5.11)
Теория активных соударений позволила наметить путь теоретического расчета константы скорости реакции: предсказала правильный ход зависимости константы скорости от температуры, позволила рассчитать для ряда реакций константы скорости по известному значению энергии активации. Однако эта теория содержит и ряд существенных недостатков, обусловленных тем, что она использует упрощенную картину действительных процессов. (Например, при подсчете числа соударений молекулы рассматриваются как сферы, что уже является упрощением, а в случае сложных молекул — очень грубым приближением). Главный недостаток теории состоит в том, что она не позволяет рассчитывать значение энергии активации исходя из параметров молекул реагирующих веществ.
5.3. ТЕОРИЯ АКТИВНОГО КОМПЛЕКСА
(ПЕРЕХОДНОГО СОСТОЯНИЯ).
Более совершенной является теория переходного состояния, в которой в качестве условия возможности протекания реакции рассматривается не столкновение двух молекул, а образование ими непрочного промежуточного комплекса, что позволяет сразу учесть необходимость определенной ориентации реагирующих молекул. Энергия системы зависит от взаимного расположения (конфигурации) атомов и сил взаимодействия между ними. Сама химическая реакция рассматривается как переход от начальной конфигурации атомов (исходные вещества) к конечной (продукты реакции) при непрерывном изменении соответствующих координат (межатомных расстояний, валентных углов). В ходе этого процесса непрерывно изменяется энергия всей системы, при этом всегда образуется некоторая промежуточная конфигурация атомов, соответствующая максимуму потенциальной энергии системы, — эту конфигурацию и называют активным (промежуточным) комплексом. Активный комплекс не является, в отличие от обычной молекулы, устойчивой частицей, так как соответствует максимуму, а не минимуму потенциальной энергии.
Рассмотрим с позиций теории переходного комплекса реакцию между молекулой АВ и атомом С:
АВ + С Û [ А... В... С ]¹ ® А + ВС.
Данную реакцию формально можно представить в виде двух последовательных стадий: разрыва связи А - В и образования связи В - С. В действительности эти стадии не происходят по отдельности: атом С при сближении с молекулой АВ начинает взаимодействовать с атомом В, в результате связь А - В слабеет (расстояние между атомами А и В увеличивается), при дальнейшем усилении связи В - С связь А - В все более слабеет до тех пор, пока не образуется молекула ВС (атомы А и В находятся на таком расстоянии, что практически не взаимодействуют друг с другом).
Изменение энергии системы при переходе от исходных к конечным веществам представлено на рис. 9. По оси абсцисс отложена “реакционная координата” (“путь реакции”), характеризующая продвижение системы по пути реакции. Энергия переходного комплекса выше энергии исходных веществ на величину Δ Е 0, которая представляет собой энергию активации реакции (для того, чтобы реакция могла произойти, система должны преодолеть потенциальный барьер Δ Е 0).
 |
Рис. 9. Изменение потенциальной энергии вдоль координаты реакции.
Согласно теории переходного комплекса между исходными веществами и комплексом устанавливается равновесие. Константа этого равновесия равна
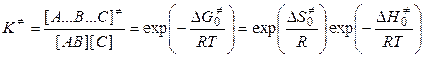 , (5.12)
, (5.12)
где 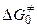 ,
,  и
и 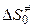 — свободная энергия, энтальпия и энтропия образования переходного комплекса из исходных веществ,
— свободная энергия, энтальпия и энтропия образования переходного комплекса из исходных веществ,
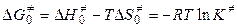 .
.
Скорость образования конечного продукта ВС можно выразить двояким образом. Во-первых, как скорость бимолекулярной реакции
 . (5.13)
. (5.13)
Во-вторых, как скорость разложения переходного комплекса (концентрация переходного комплекса выражена из ур-я (11.12)):
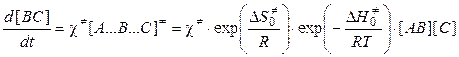 , (5.14)
, (5.14)
где c¹ — константа скорости распада переходного комплекса. Тогда
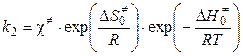 . (5.15)
. (5.15)
Чтобы получить окончательное выражение для константы скорости бимолекулярной реакции, необходимо знать константу скорости распада переходного комплекса. Полагают, что эта константа равна частоте колебаний разрывающейся “критической” связи в комплексе (в данном случае связи А - В). Подробная теория переходного состояния дает для этой частоты величину kT / h, где k — константа Больцмана, h — постоянная Планка. Тогда
 , (5.16)
, (5.16)
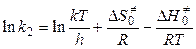 . (5.17)
. (5.17)
С другой стороны, по уравнению Аррениуса
 .
.
Тогда
 ,
,  . (5.18)
. (5.18)
Значение ур-я (5.18) состоит в том, что оно объясняет причины существования низких величин предэкспоненциального множителя, для учета которых в теории активных столкновений молекул вводится стерический фактор Р
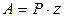 .
.
Действительно, по ур-ю (11.18) величина А зависит от энтропии образования переходного комплекса. В рассматриваемом случае образование одной частицы активного комплекса из двух частиц (молекулы АВ и атома С) должно сопровождаться уменьшением энтропии, т. е.
< 0, 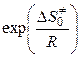 < 1.
< 1.
Так, например, если = -25 Дж/(моль·К), то
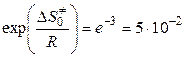 ,
,
что делает понятным появление малых стерических множителей в теории активных столкновений. С позиций теории переходного состояния низкие скорости конденсации паров обусловлены, главным образом, именно этим энтропийным фактором (при конденсации энтропия вещества значительно уменьшается).
5.4. КАТАЛИЗ
5.4.1. Основные положения катализа.
Под катализом понимают процесс изменения скорости химической реакции под действием веществ, многократно взаимодействующих с молекулами исходных реагентов с образованием промежуточных продуктов и не расходующихся в реакциях. Эти вещества называют катализаторами. Катализаторы расходуются на промежуточных этапах реакции и полностью выделяются по завершении реакции в химически неизменном виде.
Явление катализа имеет огромное значение как в природе, так и в химической промышленности. Многие природные процессы протекают под воздействием природных билогических катализаторов — ферментов, зачастую во много раз превосходящих по активности синтетические неорганические катализаторы. В промышленности, подбирая катализаторы, можно проводить химические процессы в желаемом направлении и с необходимой скоростью. Большое промышленное значение имеют газовые реакции на твердых катализаторах, например, такие как синтез аммиака из водорода и азота на железных катализаторах, содержащих добавки оксидов некоторых металлов.
Существует положительный (скорость реакции увеличивается) и отрицательный (скорость реакции уменьшается) катализ. Частным случаем положительного катализа является автокатализ, при котором свойствами катализатора обладает один или несколько продуктов реакции. Различают гомогенный и гетерогенный катализ. При гомогенном катализе все реагирующие вещества и катализатор находятся в одной фазе, при гетерогенном — катализатор составляет отдельную, обычно твердую, фазу.
Основные положения катализа:
1). Катализатор активно образует промежуточные соединения либо с одним из участников реакции, либо со всеми реагентами.
2). Катализатор в ходе реакции не изменяется химически, но может изменять свое физическое состояние (явления спекания, разрыхления катализаторов и т. д.).
3). Участие катализатора не отражается на стехиометрии реакции, однако ничтожно малое количество катализатора может значительно увеличить скорость реакции. Например, для большинства гомогенных каталитических реакций зависимость константы скорости от концентрации катализатора описывается уравнением
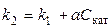 , (5.19)
, (5.19)
где k 1 —константа скорости некаталитической реакции, k 2 — константа скорости в присутствии катализатора, а — опытный коэффициент пропорциональности, С кат. — концентрация катализатора.
4). В системах из веществ, которые могут реагировать с образованием различных продуктов, введение катализатора часто способствует протеканию преимущественно или даже исключительно только одной из возможных реакций. Эта способность катализатора ускорять только одну из нескольких возможных реакций, носит название избирательного, или селективного, действия. Избирательность катализаторов имеет большое значение при промышленном осуществлении каталитических процессов.
5). При положительном катализе катализатор уменьшает энергию активации реакции и изменяет стерический фактор Р. Это связано с тем, что энергии активации образования провежуточных комплексов катализатора с исходными реагентами и образования из них продуктов реакции обычно значительно ниже энергии активации некаталитической реакции.
 |
На рис. 10 представлено изменение энергии системы в каталитической и некаталитической (бимолекулярной) реакции А + В ® С.
Рис. 10. Изменение потенциальной энергии в каталитической
и некаталитической реакциях.
В присутствии катализатора (К) эта бимолекулярная реакция протекает в две стадии с образованием промежуточного комплекса АК:
А + К ® АК,
АК + В ® С + К.
Обе стадии каталитической реакции требуют затраты энергии активации Е 1 и Е 2, однако каждая из этих энергий меньше энергии активации некаталитической реакции. Если считать, что изменение стерического фактора реакции под действием катализатора незначительно, то влияние понижения энергии активации на скорость реакции можно оценить по следующему уравнению:
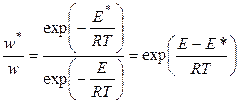 , (5.20)
, (5.20)
где w * и w — скорости каталитической и некаталитической реакции, соответственно; Е * и Е — соответствующие энергии активации. Так, при понижении энергии активации на 10 кДж/моль при 300 К скорость реакции увеличивается в 55 раз.
6). Катализатор в обратимой реакции не смещает равновесия, а в равной степени ускоряет и прямую, и обратную реакцию. В присутствии катализатора состояние равновесия достигается намного быстрее, однако он не влияет на величину константы равновесия реакции и не может увеличить выход продуктов реакции при заданных условиях. Так, для обратимой реакции А + В Û С константа равновесия равна
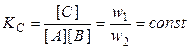 (5.21)
(5.21)
и является постоянной величиной при заданной температуре (концентрации веществ равновесные, w 1 и w 2 — скорости прямой и обратной реакций.
7). Катализаторы могут быть чувствительны к наличию в системе некоторых веществ, не участвующих в реакции. Причем возможны два случая: в первом эти вещества увеличивают активность катализатора (тогда их называют промоторами), во втором — уменьшают или вообще прекращают его действие (ингибиторы, каталитические яды).
5.4.2. Гомогенный катализ.
Каталитическим действием в гомогенной жидкой или газообразной фазе могут обладать нейтральные молекулы, положительные или отрицательные ионы, координационные комплексы и высокомолекулярные соединения (например, ферменты). Рассмотрим бимолекулярную реакцию
А + В ® [ A... В ]¹ ® D.
В отсутствие катализатора эта реакция протекает через образование переходного комплекса [ A... В ]¹ с последующим его превращением в вещество D. При введении катализатора (К) в систему возможны два случая:
1). Катализатор образует промежуточный комплекс с обоими исходными реагентами:
А + В + К 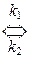 [ АВК ]¹
[ АВК ]¹ 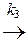 D + К.
D + К.
При этом, если выполняется условие k 1 < < k 2 < k 3, то промежуточный комплекс [ АВК ]* носит название комплекса Аррениуса и концентрация его мала (образующийся комплекс быстро либо распадается на исходные вещества, либо превращается в продукт реакции с выделением катализатора). Поскольку лимитирующей стадией реакции в данном случае является первая (образование комплекса), то в общем случае скорость реакции описывается следующим уравнением
w = k 1· C A· C B· C К = k ¢ · C A· C B, (11.22)
где С А, С В и С К — концентрации А, В и катализатора, k ¢ = k 1· C К — опытная константа скорости реакции (при условии, что С К = const).
2). Катализатор образует промежуточный комплекс только с одним из реагентов, при взаимодействии промежуточного комплекса с другим реагентом образуется продукт и выделяется катализатор:
А + К [ АК ]¹
[ АК ]¹ + В 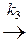 D + К.
D + К.
При этом, если выполняется условие k 1» k 2 > k 3, то промежуточный комплекс [ АК ]* носит название комплекса Вант-Гоффа. Для первой обратимой стадии образования комплекса в состоянии равновесия можно записать:
 , (5.23)
, (5.23)
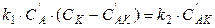 , (5.24)
, (5.24)
 , (5.25)
, (5.25)
где 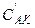 и
и  — равновесные концентрации промежуточного комплекса и реагента А, С К — общая концентрация катализатора (разность
— равновесные концентрации промежуточного комплекса и реагента А, С К — общая концентрация катализатора (разность 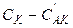 соответствует концентрации не вступившего в реакцию катализатора). Поскольку самой медленной, т.е. лимитирующей, стадией данной каталитической реакции является вторая, то скорость всей реакции (скорость образования вещества D) равна:
соответствует концентрации не вступившего в реакцию катализатора). Поскольку самой медленной, т.е. лимитирующей, стадией данной каталитической реакции является вторая, то скорость всей реакции (скорость образования вещества D) равна:
 . (5.26)
. (5.26)
Рассмотрим два предельных случая:
а) Если  , то
, то
 ,
,
где k ¢ — опытная константа скорости реакции (при условии, что С К = const). Следовательно, реакция будет протекать по уравнению реакции второго порядка.
б) Если  , то
, то
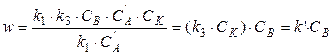
Реакция будет протекать по уравнению первого порядка и зависеть только от концентрации вещества В.
Примером гомогенного катализа в жидкой фазе является кислотно-основный катализ в растворах, протекающий в общем случае под действием различных пар частиц кислота–основание, при этом роль основания очень часто играет растворитель (например, щелочной гидролиз сложных эфиров).
Примером гомогенного катализа в газовой фазе, использующегося в химической промышленности, может служить процесс окисления оксида серы (IV) SO2 в оксид серы (VI) SO3 под действием оксидов азота (II, IV):
SO2 + NO2 ® SO3 + NO
NO + 1/2O2 ® NO2.
Суммарная реакция SO2 + 1/2O2 ® SO3.
5.4.3. Гетерогенный катализ.
Гетерогенный катализ имеет исключительно большое значение в современной химической промышленности, по некоторым оценкам до 70% химических продуктов производится с использованием гетерогенно-каталитических процессов. В качестве примеров можно привести синтез серной кислоты, основанный на каталитическом окислении оксида серы (IV) SO2 в оксид серы (VI) SO3 на платиново-ванадиевых катализаторах (Pt, V2O5); синтез аммиака из водорода и азота на железных катализаторах, промотированных оксидами калия, алюминия и других металлов; окисления аммиака в оксиды азота на платиновых катализаторах (используется в производстве азотной кислоты).
Так как при гетерогенном катализе катализатор представляет собой отдельную фазу и каталитические процессы происходят на его поверхности (границе раздела фаз), то огромную роль в протекании гетерогенно-каталитических реакций играют процессы массопереноса, т. е. для них характерны все те особенности, которые рассматривались при изучении гетерогенных химических реакций. Однако в настоящее время считается твердо установленным, что каталитическое действие твердых веществ происходит через стадию химического взаимодействия реагирующих веществ с поверхностью катализатора или отдельными ее участками, получившими название активных центров. Адсорбированные молекулы подвергаются превращению или сами по себе или же в результате взаимодействия с другими, также адсорбированными молекулами. В некоторых случаях каталитическая реакция может происходить в результате соударений молекул из газовой фазы с адсорбированными на поверхности катализатора молекулами.
Скорости процессов адсорбции (под адсорбцией понимают процесс самопроизвольного увеличения концентрации вещества на границе раздела фаз по сравнению с его концентрацией в объеме) различных веществ на различных адсорбентах изменяются в широких пределах. В общем случае различают:
1) физическую адсорбцию, обусловленную силами межмолекулярного взаимодействия, возникающими между молекулами или атомами на поверхности твердой фазы и молекулами веществ;
2) химическую адсорбцию (или хемосорбцию), обусловленную силами, близкими к химическим, т.е. хемосорбция завершается актом химического взаимодействия адсорбируемой молекулы с поверхностью твердой фазы.
При физической адсорбции количество адсорбируемого вещества при постоянном давлении уменьшается с повышением температуры. Скорость же физической адсорбции мало зависит от температуры, так как в основном определяется скоростью диффузии. При хемосорбции количество адсорбированного вещества также уменьшается с ростом температуры, однако обычно количество хемосорбированного вещества больше, чем количество физически адсорбированного вещества, а скорость процесса существенно зависит от температуры (растет по экспоненте с повышением температуры) и характеризуется определенной и значительной (от 40 до 90 кДж/моль) энергией активации. Химическая адсорбция такого типа получила название активированной.
Очень часто для одного и того же вещества, но в различных интервалах температур, можно наблюдать оба типа адсорбции (рис. 11). При низких температурах наблюдается физическая, при высоких — химическая (активированная) адсорбция. Оба эти процесса разделены промежуточной областью, которая характеризуется увеличением количества адсорбированного вещества с повышением температуры, однако из-за малой скорости адсорбции равновесие в этой области практически не устанавливается.
Рис. 11. Изобара адсорбции вещества.
 |
В общем случае гетерогенно-каталитические процессы можно условно разбить на следующие стадии:
1. диффузия исходных веществ к поверхности катализатора,
2. адсорбция молекул исходных веществ на активных центрах катализатора,
3. собственно химическая реакция между адсорбированными молекулами,
4. десорбция продуктов реакции с поверхности катализатора,
5. отвод продуктов реакции вглубь раствора.
Поскольку гетерогенно-каталитические реакции происходят между адсорбированными молекулами, то в уравнение скорости таких реакций входят поверхностные концентрации реагирующих веществ. Так, например, гидрирование этилена на медном катализаторе происходит между адсорбированными молекулами этилена и водорода:
С2Н4 (адс.) + Н2 (адс.) = С2Н6,
а скорость этой реакции пропорциональна произведению поверхностных концентраций:
w = k ·[С2Н4 (адс.)]·[Н2 (адс.)].
5.4.4. Активные центры на поверхности катализатора.
В настоящее время считается доказанным, что лишь небольшая часть поверхности катализатора участвует в каталитическом процессе. Те участки поверхности, которым присуща каталитическая активность, получили название активных центров. Существование активных центров следует прежде всего из явления отравления катализаторов — при проведении каталитической реакции исходные вещества должны быть тщательно очищены от примесей некоторых веществ (каталитических ядов), способных адсорбироваться на поверхности катализатора и резко снижать его активность. Опыты показывают, что количество этих веществ, достаточное для покрытия всего 10-15% поверхности катализатора, может снижать его каталитическую активность более чем на 70%. Другим фактом, свидетельствующем от неоднородности поверхности катализатора, является изменение теплоты адсорбции газа на поверхности в зависимости от величины ее заполнения. Как правило, первые порции газа адсорбируются с выделением большей теплоты, чем последующие.
Вопрос о природе активных центров составляет предмет теории каталитического действия твердых веществ. Исторически первой была теория Х. Тэйлора, согласно которой каталитически активные центры представляют атомы или группы атомов, выступающие над поверхностью катализатора. Более детальное рассмотрение природы активных центров дано в мультиплетной теории А.А. Баландина (слово “мультиплетный” может быть переведено как составной, сложный) и в теории каталитически активных ансамблей Н.И. Кобозева.
По мультиплетной теории молекулы реагирующих веществ образуют химические связи с несколькими атомами катализатора, входящими в активный центр. В простейшем случае в активный центр входят два атома, образующие “дублет”. В данной теории предполагается, что атомы дублета или другого мультиплета составляют осколок кристаллической решетки металла, катализирующего реакцию. Это важная конкретизация строения активного центра, так как она позволяет установить взаимное расположение атомов металла в мультиплете и расстояния между ними.
В отличие от мультиплетной теории теория каталитически активных ансамблей предусматривает возможность существования активных центров из атомов, не входящих в кристаллическую решетку. Она допускает также существование активных центров всего из одного атома металла. Характерной чертой данной теории является предположение о том, что атомы активных центров образуют “аморфную” фазу на поверхности носителя или на поверхности кристаллических граней самого металла.
|
|