Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Классификация каталитических процессов
|
|
Все реакционные системы принято делить на гомофазные и гетерофазные. В первом случае в реакционной системе отсутствуют границы раздела фаз. Гетерофазные реакционные системы имеют хотя бы одну границу раздела фаз. Каталитические процессы классифицируют следующим образом.
1. Гетерогенно-каталитические процессы.
2. Гомогенно-каталитические процессы.
3. Микрогетерогенный катализ.
А. Процессы мицеллярного катализа.
В. Процессы, катализируемые микрочастицами (например, наночастицами металлов) в растворах.
4. Межфазный катализ.
5. Ферментативный катализ.
Если катализатор и реагенты находятся в одной фазе в молекулярно-дисперсном состоянии и в объеме этой фазы протекают реакции, это гомогенно-каталитический процесс. Гомогенные каталитические процессы могут протекать как в гомофазных (фаза, в которой протекает процесс, единственная в системе), так и в гетерофазных системах. Если в системе присутствуют другие фазы, кроме той, в которой протекает процесс, то это гомогенно-каталитический процесс в гетерофазной системе. Например, гидрирование в жидкой фазе с участием растворенного в жидкой фазе катализатора – это гомогенно-каталитический процесс в гетерофазной системе, т.к. есть граница раздела фаз – Н2/раствор, а реакция протекает в жидкой фазе, в которой растворен водород. Реакция этерификации этанола уксусной кислотой в присутствии растворенного кислотного катализатора при отсутствии расслаивания является гомогенно-каталитическим процессом в гомофазной системе. Гомогенно-каталитический процесс может протекать в газофазной системе – окисление сернистого ангидрида в серный ангидрид кислородом при катализе диоксидом азота.
Если процесс протекает на поверхности твердого катализатора, это гетерогенно-каталитический процесс. Гетерогенно-каталитические процессы всегда протекают в гетерофазных системах, поскольку есть, по крайней мере, одна граница раздела фаз – поверхность твердого катализатора, а реагенты, продукты и другие компоненты реакционной системы могут быть газообразными, жидкими и твердыми.
Микрогетерогенный катализ, являющийся промежуточным между гомогенным и гетерогенным, объединяет мицеллярный катализ и катализ нанокластерами металлов и коллоидными металлсодержащими системами.
Мицеллярный катализ. Мицеллы – ассоциаты поверхностно-активных веществ (ПАВ), образующиеся самопроизвольно в растворителе. Существует критическая концентрация мицеллообразования (ККМ), ниже которой ПАВ образуют истинные растворы, а при концентрации выше ККМ весь избыток ПАВ находится в виде мицелл, содержащих 50-100 молекул. Молярная масса мицеллы – 103-105 Д.
Мицеллярный катализ, т.е. ускорение химических реакций в присутствии ПАВ объясняют, в первую очередь, концентрированием реагентов в мицеллах, а также влиянием условий в мицелле на величины констант скорости и диссоциации распадающихся на ионы реагентов. В ряде систем ускорение достигает 1000 раз при переходе к реакциям в мицеллах. В условиях мицеллярного катализа могут протекать реакции, катализируемые кислотами, основаниями, нуклеофилами, металлокомплексами и ферментами. Существует ряд математических моделей для описания таких процессов.
Межфазный катализ. При проведении химических процессов в гетерофазных системах, включающих несмешивающиеся жидкие фазы или твердую фазу, нерастворимую в используемых жидких фазах, целевая реакция может быть значительно ускорена за счет применения катализаторов межфазного переноса. Одна из фаз содержит раствор исходного реагента (субстрата) в органическом растворителе (иногда реагент и является жидкой фазой), а другая жидкая фаза является водным раствором щелочи или нуклеофила или твердым основанием (щелочью). Поскольку фазы не смешиваются (взаимная растворимость очень мала), то субстрат не контактирует с основанием или нуклеофилом, и реакция практически не идет. Участие катализатора межфазного переноса обеспечивает перенос реагирующих молекул и продуктов реакций между фазами и снижает энергию активации целевой реакции.
В качестве катализаторов межфазного переноса в жидкофазных системах чаще всего используют четвертичные аммониевые или фосфониевые соли. Например, бензилтриэтиламмонийхлорид (ТЭБАХ), тетрабутиламмонийбромид (ТБАБ), тетрабутиламмонийхлорид (ТБАХ), метилтриоктиламмонийхлорид (аликват 336), тетрабутиламмонийгидросульфат (ТБАГС), бензилтриметиламмонийхлорид (ТМАБХ), трибутилгексадецилфосфонийбромид (ТБГДФБ), тетрафениларсонийхлорид и др. Возможно применение разнообразных аминов и диаминов (N, N’- диметилпиперазин, N, N, N’, N’- тетраметилэтилендиамин, N-бутилпиперидин, триалкиламины и др.). В системах «твердая фаза-жидкость» целесообразно использовать каталитические количества краун-эфиров. В качестве катализаторов межфазного переноса могут служить и другие вещества, способные к образованию ионных пар с анионами или катионами.
Рассмотрим принцип действия катализатора межфазного переноса в системе «жидкость-жидкость» на самом простом примере. Пусть необходимо провести гидролиз галоидного арила для получения замещенного фенола. Процесс должен протекать в двухфазной системе «водная щелочь-ароматика», так как щелочь необходима для связывания выделяющейся кислоты. Без катализатора межфазного переноса реакция не происходит из-за
ArHal + NaOH = ArOH + NaHal
отсутствия взаимной растворимости. При введении в систему вещества, содержащего липофильный катион (Q+X-), растворяющийся в обеих фазах, в водной фазе протекает обмен анионами межфазного катализатора со щелочью. Далее липофильный катион Q+ переносит
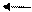
 Q+X- + NaOH Q+OH + NaX-
Q+X- + NaOH Q+OH + NaX-
гидроксил в органическую фазу:

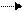 Q+OH(водная фаза) Q+OH(органическая фаза)
Q+OH(водная фаза) Q+OH(органическая фаза)
В органической фазе происходит обмен анионами (нуклеофильное замещение) с образованием замещенного фенола:

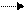 ArHal + Q+OH ArOH + Q+Hal
ArHal + Q+OH ArOH + Q+Hal
Катион Q+ образует ионную пару с уходящей группой и способствует её переносу в водную фазу. И каталитический цикл завершается регенерацией катализатора межфазного переноса.
Q+ Hal (органическая фаза) Q+ Hal (водная фаза)

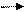 Q+ Hal + NaX- Q+X- + NaHal
Q+ Hal + NaX- Q+X- + NaHal
Несколько сложнее механизм катализа межфазного переноса в случае концентрированных систем «жидкость-жидкость» и «жидкость-твердое». С ними можно ознакомиться в ряде монографий (см. список рекомендованной литературы).
В заключение этого раздела отметим преимущества данного метода.
1. Метод позволяет исключить использование дорогих органических растворителей (гомогенизаторов).
2. Возможно значительное повышение скорости процесса, селективности и выхода целевых продуктов.
3. Метод допускает использование соединений, чувствительных к гидролизу, действию щелочей, изомеризации и пр.
4. Исключается необходимость использования щелочных металлов, их гидридов, алкоксидов, амидов с их пожароопасностью и чувствительностью к влаге.
За последние десятилетия межфазный катализ нашел широкое применение в разных областях органического синтеза.
Ферментативный катализ. Ферментативный катализ используется людьми тысячи лет, задолго до появления самого понятия «катализ». Получение молочно-кислых продуктов, сыра, приготовление теста, вина, красителей и др. продуктов включало применение ферментативных процессов. Технология этих процессов передавалась из поколения в поколение и была эмпирически отработана до совершенства. Считают, что в эволюции жизни и появлении сложных биологических систем (включая человека) важную роль сыграл ферментативный катализ.
Ферменты (энзимы) – биологические катализаторы обладают уникальными свойствами: высокой производительностью в расчете на один реакционный центр и селективностью, связанной со специфичностью действия. Работают ферменты в очень мягких условиях, при атмосферном давлении и температуре до 40о. В биологических системах отсутствуют неводные растворители и сильные кислоты и основания (рН ≈ 5-8). Например, фермент уреаза гидролизует только молекулы мочевины, не обращая внимания на другие амиды, и делает это гораздо эффективнее обычных кислотных катализаторов (табл. 2). Константы скорости для реакций, катализируемых ферментами, как правило на несколько порядков превышают константы скорости аналогичных реакций, катализируемых веществами других классов, чаще всего в гораздо более жестких условиях (табл. 2, для кислотных катализаторов).
Таблица 2
Сравнение каталитической активности ферментов и кислотных катализаторов
| Реакция и субстрат | Катализатор | Константа скорости второго порядка, л·моль-1∙ с-1 | Температура, оС |
| Гидролиз сложных эфиров Этилбензоат Этиловый эфир N-бензоил-L-тирозина | Н3О+ Химотрипсин | 9, 0∙ 10-5 1, 9∙ 104 | |
| Гидролиз аденозин-трифосфата (АТФ) | Н3О+ Миозин | 4, 7∙ 10-6 8, 2∙ 106 | |
| Гидролиз амидов Бензамид Амид N-бензоил- L-тирозина Мочевина | Н3О+ Химотрипсин Н3О+ Уреаза | 2, 4∙ 10-6 14, 9 7, 4∙ 10-6 5, 0∙ 106 |
Международные правила номенклатуры ферментов в зависимости от выполняемых ими функций выделяют шесть основных классов с соответствующими подклассами внутри каждого класса (табл.3).
Таблица 3.
Классификация ферментов
| Класс. Функция | Подклассы | Класс, функция | Подклассы |
| СН-ОН С=О СН-СН СН-NH2 CH-NH НАД(Ф)Н | 2. Транс-феразы Переносят сле-дующие груп-пы (см. под-классы) | одноуглеродные остатки остатки альдегидов и кетонов ацильные остатки гликозильные остатки алкильные (кроме СН3) и арильные группы азотистые группы фосфорсодержащие группы |
| сложные эфиры гликозидные соединения простые эфиры и тиоэфиры пептидные связи связи C-N, кроме пептидной |
| С - С С - О С - N C - S C - Hal |
| рацемазы и эпиме-разы цис-транс-изоме-разы внутримолекуляр-ные оксидоредук-тазы внутримолекуляр-ные трансферазы внутримолекуляр-ные лиазы | 6. Лигазы (синтетазы) Одновременно с расщеплени-ем АТФ обра-зуют связи (см. подклассы) | С - О С - S C - N C - C |
Приведенная таблица может помочь ориентироваться во множестве уже известных ферментов и их названиях.
Ферментом может быть глобулярный белок, в активном центре которого собраны функциональные группы, входящие в состав аминокислотных остатков этого белка. В других случаях в состав активного центра входит прочно связанная с белковой цепью простетическая группа (например, липоевая кислота) или слабо связанный кофермент (например, АТФ). Комплексы металлов также могут входить как в простетическую группу (например, железо(II) входит в состав активного центра гемоглобина), так и в кофермент (например, кобальт(III) входит в состав кофермента витамина В12). Фермент в целом называют холоферментом, а то, что остается после удаления кофермента, - апоферментом.
В соответствии с требованиями, предъявляемыми при подборе катализаторов-ферментов, их подразделяют на следующие группы:
1. Ферменты без коферментов – простые гидролазы, лиазы и изомеразы.
2. Ферменты, которые не требуют наличия кофермента (содержат прочно связанную простетическую группу, например, флавиновую или пиридоксальную) – трансаминазы, пероксидазы и т. п.
3. Ферменты, которые требуют регенерации кофермента, обычно АТФ или НАД(Ф)Н - например, киназы, большинство оксидоредуктаз.
4. Ферменты, которые встречаются в многоферментных системах.
Ферменты первой группы используются пока шире, часто и в промышленном масштабе (синтез L-аминокислот, 6-аминопеницилиновой кислоты, изомеризация глюкозы во фруктозу и т. д.). Остальные группы ферментов требуют создания особых условий и до сих пор находят применение только в лабораторных синтезах.
Что такое ферменты и за счет каких факторов они работают так эффективно?
Объяснение состоит в том, что фермент обладает способностью формировать так называемый активный центр и создавать в нем специфическое окружение, в котором протекание катализируемой реакции происходит несоизмеримо быстрее, чем в растворе.
В активном центре происходит специфическое связывание субстрата. Например, сбраживание глюкозы в спирт дрожжами требует участия более 12 ферментов, каждый из которых выполняет свою функцию. Это возможно только благодаря высокой специфичности.
Различают абсолютную специфичность – специфичность по отношению к одному конкретному субстрату (уреаза – мочевина; галактокиназа переносит фосфат от АТФ только на Д-галактозу, но не на ее стерео изомеры Д-глюкозу и Д-маннозу);
абсолютную групповую специфичность – специфичность к определенному классу субстратов (спирты, альдегиды, простые или сложные эфиры). Так, протеолитический фермент пепсин специфичен в отношении гидролиза пептидной связи. Алкогольдегидраза окисляет только спирты, а лактикодегидраза – только α -оксикислоты;
относительная групповая специфичность – фермент действует предпочтительно на один класс соединений, но может в некоторой степени действовать и на представителей других классов, превращая их с меньшими скоростями, чем представителей основного класса. Трипсин способен расщеплять как пептидные, так и сложноэфирные связи.
Оптическая специфичность – общее свойство большей части ферментов взаимодействовать с веществами, имеющими определенную оптическую активность.
Основу ферментов составляют белки, поэтому можно сказать, что ферменты – это белки, способные катализировать химические реакции. Открыты ферменты были в 30-е годы 19-го века, и примерно сто лет ушло на то, чтобы прийти к приведенному определению. Не всякий белок может быть ферментом. По внешней форме белки бывают линейные (фибриллярные) и глобулярные. Только глобулярные белки могут быть ферментами. Белки – это полипептиды, т.е. полимеры, состоящие из аминокислотных остатков, соединенных пептидной связью. Ниже показана реакция образования дипептида. Все природные белки построены из примерно 20 различных аминокислотных остатков, отличающихся строением
NH2-CH-COOH + H2N-CH-COOH → NH2-CH-CO-HN-CH-COOH + H2O


X1 X2 X1 X2
группы Х. Каталитические свойства могут проявлять полипептиды (белки), имеющие молярную массу не менее 5000.
Строение белков имеет три разных уровня.
Первичная структура определяется последовательностью аминокислотных остатков, образующих полипептидную цепь.
Вторичную структуру белка определяют дополнительные связи, возникающие между группами, принадлежащими различным аминокислотным остаткам, находящимся в разных частях полипептидной цепи. К числу таких связей относятся водородные, электростатические, координационные, гидрофобно-гидрофобные и Ван-дер-Ваальсовы взаимодействия. В результате образования дополнительных связей отдельные участки полипептидной цепи образуют α -спирали, петли и β -тяжи.
Третичная структура белка формируется в результате сворачивания отдельных участков полипептидной цепи в относительно автономные глобулярные образования, называемые доменами. Окончательное формирование третичной структуры происходит благодаря специфическим взаимодействиям, возникающим между отдельными доменами, каждый из которых сворачивается самостоятельно. Длинные полипептидные цепи обычно формируют несколько доменов, величина которых значительно варьирует, составляя в среднем 150 аминокислотных остатков. Взаимодействия между доменами приводят к образованию глобулы.
Домены характеризуются тем, что число взаимодействий между аминокислотными остатками в составе домена значительно превышает таковое между соседними доменами. Благодаря этому междоменные области оказываются сравнительно легко доступными для растворителя и содержат полости объемом 20-30 кубических ангстрем, включающие несколько молекул воды. «Архитектурные принципы» построения отдельных доменов различны, что может быть связано с выполнением ими разных функций.
Активные центры мультидоменных (в большинстве случаев – двухдоменных) ферментов, как правило, располагаются в междоменной области. Таким образом, каждый из доменов вносит свой вклад в связывание участников реакции.
Важным следствием расположения активного центра на границе между доменами является обеспечение гибкости, подвижности данной области молекулы благодаря тому, что в ходе конформационных изменений, вызываемых связыванием субстратов, домены претерпевают взаимное перемещение.
Между размером молекулы биологического катализатора (т. е. длиной его полипептидной цепи) и сложностью выполняемой им функции существует прямая зависимость. Усложнение функциональных свойств достигается как за счет формирования активного центра на границе раздела между двумя каталитическими доменами, так и за счет появления дополнительных доменов, ответственных за регуляцию активности. Такие ферменты, как лизоцим и гликогенфосфорилаза, резко различаются по размерам (129 аминокислотных остатков в первом и 842 – во втором), хотя оба катализируют реакции расщепления гликозидной связи. Функциональный смысл «утяжеления» молекулы гликогенфосфорилазы состоит в придании ей дополнительной способности координировать работу активного центра в соответствии с сигналами, поступающими из внешней среды (изменение концентраций метаболитов, нервные и гормональные сигналы).
К факторам, определяющим высокую эффективность ферментов, относят:
1. Концентрационный эффект.
2. Ориентационный эффект.
3. Полифункциональность реакционного центра.
Сущность концентрационного эффекта в случае ферментов ничем не отличается от концентрационного эффекта в гетерогенном катализе. Фермент в своем реакционном центре создает локальную концентрацию субстрата, которая существенно выше, чем средняя концентрация в растворе. В реакционном центре фермента селективно концентрируются молекулы, которые должны прореагировать между собой. Такой эффект может приводить к ускорению реакции на несколько порядков.
При протекании обычных химических реакций важно, какими частями происходит столкновение реагирующих молекул. То есть, молекулы при столкновении должны быть соответствующим образом ориентированы друг относительно друга. В реакционном центре фермента при координации молекулы субстрата и образовании фермент-субстратного комплекса происходит четкая ориентация реагирующих молекул за счет взаимодействия с функциональными группами реакционного центра. Это приводит к ускорению реакций примерно на три порядка.
Под полифункциональностью реакционного центра фермента понимают одновременное или согласованное воздействие функциональных групп, входящих в состав реакционного центра, на молекулу субстрата. При этом происходит не только фиксация превращающейся молекулы в строго определенном положении (см. предыдущий пункт), но и изменение характеристик самой молекулы: растягивание связей, изменение валентных углов. Эти изменения приводят к повышению реакционной способности субстратов, т.е., к их активации и ускорению их превращения.
Кинетика ферментативного катализа имеет некоторые особенности. Способность ферментов специфически связывать свои субстраты обусловливает важнейшую особенность катализируемых ими реакций: они начинаются с образования фермент-субстратного комплекса. Связывание субстратов ограничивает их подвижность, сближает и ориентирует их относительно друг друга оптимальным образом для осуществления реакции; уменьшение степеней свободы поступательного и вращательного движения приводит к снижению энтропии. Важным следствием сближения и взаимной ориентации реагирующих групп субстратов, с одной стороны, и функциональных групп фермента, с другой, является то, что катализ становится внутримолекулярным. Это существенно увеличивает его эффективность, так как продуктивные столкновения между молекулами в растворе относительно редки.
Рассмотрим механизм функционирования ферментативного катализатора на примере гидролитического фермента химотрипсина.
Химотрипсин – фермент поджелудочной железы, функция которого в организме заключается в расщеплении белков пищи, т.е. пептидной связи. Кроме этого химотрипсин может катализировать гидролиз сложных эфиров и некоторые другие реакции. Брутто формула химотрипсина, включающего 241 остаток аминокислот, не несет информации о строении: С1105H1732O344N300S12, также как перечисление количества аминокислотных остатков: аланин22аргинин3аспарагиновая кислота8аспарагин14глутаминовая кислота3 глутамин10глицин24гистидин2изолейцин10лейцин19лизин14метионин2полуцистин10пролин9 серин28треонин22триптофан8тирозин4валин23фенилаланин6. Перечисленные аминокислотные остатки соединены в полипептидную цепь в определенной последовательности (первичная структура). Отдельные части полипептидной цепи за счет образования дополнительных связей (см.выше) скручиваются в α -спирали, β -тяжи и петли (вторичная структура). Перечисленные элементы вторичной структуры за счет дополнительных взаимодействий сворачиваются в два домена, в месте соприкосновения которых возникает активный центр фермента, включающий остаток серина (Х – -СН2ОН), аспарагиновой кислоты (Х - -СН2СОО-), гистидина
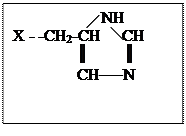
Механизм реакции гидролиза, например, сложного эфира с участием химотрипсина показан на схеме (рис. 2). При подходе субстрата к активному центру фермента неполярная гидрофобная часть субстрата взаимодействует с гидрофобной частью активного центра (на схеме не показано), протон от кислорода серина переходит на азот гистидина, а протон от второго азота гистидина смещается к аниону остатка аспарагиновой кислоты. Образовавшийся из гидроксильной группы серина сильный нуклеофил - -О- атакует электрофильный углерод субстрата, в то время как нуклеофильная часть субстрата взаимодействует с протоном, связанным с азотом гистидина. В результате этих взаимодействий образуется фермент-субстратный комплекс. На следующей стадии рвется связь С-Х в субстрате, уходит молекула НХ (ROH), а ее место в активном центре занимает молекула воды. Протон от остатка аспарагиновой кислоты возвращается к второму азоту гистидина. Затем рвется предварительно активированная связь О-Н в молекуле воды (протон связывается с первым азотом гистидина, а гидроксил – с углеродом бывшего субстрата). Протон от второго азота гистидина опять возвращается к остатку аспарагиновой кислоты. И наконец выделяется кислота, место которой занимает новый субстрат или активный центр возвращается в исходное состояние (рис. 2).
|
|