Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Задание для самоконтроля
|
|
1. Изобразите схематически глобальный цикл элемента азота, обозначьте в нем процессы превращения азотсодержащих соединений, резервуары (пулы) и потоки азота.
2. Проанализируйте, какие процессы приводят к накоплению элемента азота в различных резервуарах (пулах). Приведите примеры таких природных и антропогенных процессов и их последствий.
3. Приведите примеры процессов превращения одних соединений азота в другие, применяющиеся в хозяйственной деятельности человека.
4. Приведите примеры негативного воздействия соединений азота на человека и окружающую среду.
В атмосферу выбрасывается большое количество химических веществ, находящихся в газовом и аэрозольном состояниях. Здесь они претерпевают ряд физико-химических изменений за счет механизмов фотохимической трансформации, нуклеации, конденсации/испарения и коагуляции. Все эти механизмы взаимосвязаны между собой.
Наиболее распространенными загрязнителями, сопутствующими процессам сгорания топлива являются оксид азота (I) (N2O), оксид азота (II) (NO), и диоксид азота (NO2). Оксид азота (I), часто используемое анестезирующее средство, известное как “веселящий газ”, в природе образуется в результате микробиологических процессов и является естественной составляющей атмосферы с содержанием приблизительно 0.3 ppm.
В нормальных условиях оксид азота представляет собой бесцветный газ без запаха, диоксид азота – бурый газ с резким запахом. Суммарно антропогенные источники выбрасывают в атмосферу почти 100 миллионов тонн оксидов азота в год, что на порядок превышает эмиссию естественных источников NOx.
Оксиды азота физиологически активны. Так, N2O — средство для наркоза, в высоких концентрациях вызывает удушье. Другие оксиды азота сильно ядовиты: NO действует на центральную нервную систему, в больших концентрациях превращает оксигемоглобин в метгемоглобин; NO2 и N2O4 разрушающе действуют на легкие, в тяжелых случаях вызывают отек, понижают кровяное давление. При длительной работе в атмосфере, содержащей эти оксиды, развиваются различные хронические заболевания; ПДК NO2 9 мг/м3, остальных оксидов азота — 5 мг/м3 (в пересчете на NO2).
Негативное воздействие оксида азота отмечается не только для человека, но и для растений. В лабораторных условиях концентрации NO2, порядка единиц ppm, приводят к разрушению растительных тканей и появлению пятен на листьях, более высокие концентрации (10 ppm) вызывают понижение уровня фотосинтеза в хлоропластах.
В выбросах промышленных предприятий содержится в среднем от 50 до 1000 ppm оксидов азота.
Поступая в атмосферу оксиды азота вступают в химические реакции с гидроксилом, кислородом, озоном. Поэтому в выбросах автотранспорта, факелах ТЭЦ и других объектах, часто обнаруживаются вещества, которые не присущи первичным промышленным выбросам, например пероксинитраты, органические серо- и азотсодержащие соединения, органические кислоты и пероксиды. Большая часть химических реакций в атмосфере инициируется светом, зависит от интенсивности и спектрального состава солнечного света. Накопление в атмосфере продуктов реакций зависит не только от физико-химических условий: температуры, давления, солнечного спектра, но и от соотношения скоростей химической реакции и переноса (рассеивания) веществ.
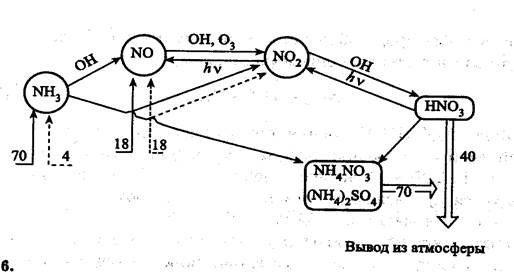
Рис. ХХХ. Атмосферный цикл соединений азота (числа –млн т элементного азота в год.
Соединения азота в тропосфере – в основном это оксиды азота, аммиак, соли аммония, азотная кислота и нитраты. Из оксидов азота следует выделить N2O, NO и NO2. Другие оксиды азота в условиях тропосферы являются неустойчивыми.
N2O - поступает в атмосферу в основном в результате протекания процессов денитрификации.
Выводится N2O из атмосферы в основном в процессах фотодиссоциации:
N2O + hν = N2 + O, λ < 250 нм (63)
N2O + O(1D) = N2 + O2, (64)
N2O + O(1D) = 2NO (65)
Константы скорости реакций (64) и (65) при 298 К равны 7, 4∙ 10-11 и 8, 6∙ 10-11см3с-1.
В тропосфере NO взаимодействует с гидропероксидными радикалами:
NO + O3 = NO2 + OH·
NO + O3 = NO2 + O2
Диоксид азота в тропосфере разлагается:
NO2 + hν = NO + O(3P), λ < 398 нм
В атмосферном цикле соединений азота важной составляющей является образование азотной кислоты:
NO2 + OH· = HNO3
Часть азотной кислоты разлагается:
HNO3 = NO2 + OH· или
HNO3 + OH· = NO3 + H2О
Основное количество азотной кислоты выводится из тропосферы с атмосферными осадками в виде растворов HNO3 и её солей.
Ежегодно из природных источников в атмосферу поступает около 70 млн т NH3 в пересчете на азот. Антропогенный вклад в загрязнение атмосферы аммиаком составляет около 4 млн т. Часть аммиака вступает во взаимодействие с радикалами (в основном с гидроксильным радикалом):
NH3 + OH· = NH2 + H2О
В дальнейшем NH2 легко окисляется до оксида азота.
Основные понятия химической кинетики
Химическая кинетика изучает скорость и молекулярный механизм химических реакций. В химической кинетике используются как методы квантовой механики, молекулярной статистики и термодинамики, так и свои специфические методы. Задачи химической кинетики – установить зависимость между строением, энергетическими характеристиками химических связей и реакционной способностью веществ, изучить влияние различных факторов на скорость и механизм химических реакций.
Для осуществления химического процесса необходимо преодоление энергетического барьера – перевода реагентов в активное состояние. Энергия, необходимая для приведения реагирующих частиц в реакционно-способное состояние называется энергией активации. Это понятие введено С. Аррениусом и Я. Вант-Гоффом. Величина энергии активации отражает время протекания химических реакций – доли секунды или геологические периоды.
Скорость реакции – это число элементарных актов реакции, происходящих в единицу времени. В ходе реакции с изменением условий протекания скорость изменяется, поэтому вводят понятие мгновенной или истинной скорости.
Мера скорости мгновенной реакции –бесконечно малоеизменение количества (или концентрации) реагента или продукта реакции во времени:
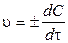 ,
,
где u – скорость химической реакции, моль/дм3× с; С – концентрация реагентов или продуктов, моль/дм3;  – время изменения концентрации, с. Скорость всегда положительна, поэтому вводят знак ±, в зависимости от того как определяется скорость по исходным веществам (знак -) или по продуктам реакции (знак +).
– время изменения концентрации, с. Скорость всегда положительна, поэтому вводят знак ±, в зависимости от того как определяется скорость по исходным веществам (знак -) или по продуктам реакции (знак +).
Различают простые (элементарные) и сложные реакции. В простых реакциях уравнение отражает механизм.
Сложные реакции состоят из нескольких стадий, описываемых простыми реакциями и суммарное уравнение не отражает их механизм. Так, например, реакция разложения 2N2O5(г) → 4NO2(г) + O2(г) протекает в несколько ступеней:
I ступень: N2O5(г)↔ NO2(г) + NO3(г) (быстро);
II ступень: NO2(г) +NO3(г) ↔ NO2(г) + NO (г) + O2(г) (медленно);
III ступень: NO (г) +NO3(г) ↔ 2NO2(г) (быстро).
Сложные реакции могут быть параллельными, последовательными, сопряжёнными, цепными.
По агрегатному состоянию реагентов все химические реакции можно систематизировать следующим образом (табл. 1).
Таблица 1
| Химические реакции | Фазовое состояние | Агрегатное состояние | Направление реакции |
| гомогенные | однофазные | жидкое твердое газообразное | обратимые uпр @ uобр |
| необратимые uпр > > uобр | |||
| гетерогенные | многофазные | жидкость – газ жидкость – твердое тело газ – твердое тело жидкость – газ – твердое тело | обратимые uпр @ uобр |
| необратимые uпр > > uобр |
Большинство химических процессов являются обратимыми в той или иной степени, для которых скорости прямой и обратной реакций соизмеримы, процесс заканчивается наступлением равновесия, а в системе присутствуют как продукты реакции, так и исходные вещества.
Получение экспериментальных данных по скоростям химических реакций позволили Н.Н. Бекетову в 1864 г. высказать гипотезу о количественной взаимосвязи между массами реагентов и временем протекания реакции.
В 1867 г. голландцы К. Гульдберг и П. Вааге открыли основной закон химической кинетики – закон действия масс: при постоянной температуре скорость химической реакции прямо пропорциональна произведению мгновенных концентраций, возведенных в степени их стехиометрических коэффициентов.
При этом стехиометрические коэффициенты должны отражать механизм элементарных актов реакции.
Кинетическое уравнение, отражающее механизм взаимодействия, может быть получено только в результате экспериментального изучения реакций и не всегда совпадает с суммарным стехиометрическим уравнением.
Для реакции в общем виде: 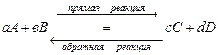 , формальное условное кинетическое уравнение прямой реакции
, формальное условное кинетическое уравнение прямой реакции 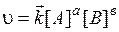 , обратной –
, обратной – 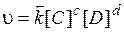 . Вид кинетического уравнения зависит от порядка реакции.
. Вид кинетического уравнения зависит от порядка реакции.
В общем случае постулат химической кинетики для реакции можно выразить в общем виде как 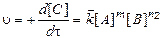 , где коэффициенты n 1, n 2, определяют порядок реакции по данному компоненту (коэффициенты n могут быть целочисленными, дробными или равными нулю.
, где коэффициенты n 1, n 2, определяют порядок реакции по данному компоненту (коэффициенты n могут быть целочисленными, дробными или равными нулю.
Порядок реакции можно определить графически, используя метод избыточных концентраций, или интегральными способами, основанными на уравнениях кинетических кривых (рис. 1, 2).
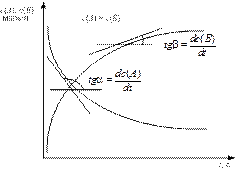
Рис. 1. Кинетические кривые, отражающие изменение концентрации реагентов (А) и продуктов реакции (В) во времени.
Скорость реакции зависит от нескольких факторов:
— от природы реагентов (типа химической связи, размеров и формы молекул, агрегатного состояния веществ, природы растворителя для реакций. протекающих в растворах);
— от концентрации реагентов или парциального давления газообразных веществ;
— от поверхности соприкосновения реагирующих веществ для гетерогенных реакций;
— от температуры;
— от присутствия катализаторов или других веществ.
Коэффициент пропорциональности k в кинетическом уравнении называется константой скорости и определяет скорость, не зависящую от концентрации.
Физическую сущность k раскрывает зависимость, установленная в 1884 г. Я.Вант-Гоффом из молекулярно-кинетических представлений для элементарных химических реакций, известная как уравнение Аррениуса:
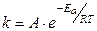 ,
,
где предэкспоненциальный множитель А – постоянная Аррениуса, определяющая число молекул, участвующих в реакции, может быть вычислена теоретически из молекулярно-кинетической теории или статистической термодинамики; Еа – по С.Аррениусу (1889 г.) энергия активации, ранее рассматривалась в химической кинетике как эмпирическая постоянная, не зависящая от температуры. В настоящее время появилась возможность ее приближенной оценки с помощью методов квантовой химии; R – универсальная газовая постоянная, 8, 313 Дж/(моль× К); Т – термодинамическая температура, К.
Влияние на скорость реакции температуры С.Аррениус трактовал как сдвиг равновесия между неактивными и активными молекулами. (Основной вклад С.Аррениуса в развитие химической кинетики состоит во введении представлений об энергии активации как потенциальном барьере, который должны преодолеть реагирующие молекулы прежде, чем стать продуктами реакции). Размерность константы скорости зависит от порядка реакции, который определяют экспериментально. Порядок реакции может быть 0-м, 1-м, 2-м, 3-м или дробным и определяет форму кинетического уравнения, отражающего механизм процесса.
Схема определения порядка реакции:
| А ® продукт реакции | ||
| Постройте зависимость [ А ] от времени (t). Является ли она линейной? | Да ® | Реакция имеет нулевой порядок по А: 
|
| ¯ НЕТ | ||
| Реакция имеет ненулевой порядок по А: определите скорость реакции по тангенсам наклона касательной в нескольких точках кинетической кривой [ А ]–t. Постройте зависимость u = f [ А ]. Является ли она линейной? | Да ® | Реакция имеет первый порядок по А: 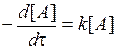
|
| ¯ НЕТ | ||
| Реакция имеет порядок, отличный от нулевого и первого: определите скорость реакции по тангенсам угла наклона касательной к кривой [ А ]–t. Постройте зависимость u = f [ А ]2. Является ли она линейной? | Да ® | Реакция имеет второй порядок по А: 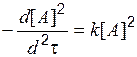
|
| ¯ НЕТ | ||
| Реакция имеет порядок, отличный от нулевого, первого и второго |
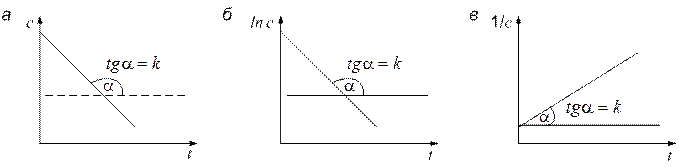
Рис. 2. Схема графического метода определения порядка реакции: а – 0-й порядок; б – 1-й порядок; в – 2-й порядок
Кинетические уравнения скорости отражают механизм протекания и позволяют определить мгновенную (дифференциальная форма) скорость в заданный момент време- ни при известной концентрации исходного вещества (табл.2).
Таблица 2
Кинетические уравнения скорости химических реакций;
с – содержание исходного вещества (с0 – начальная концентрация исходного вещества)
| Порядок реакции | Кинетические уравнения | Период полураспада, τ 1/2 | Единицы измерения скорости | |
| Дифференциальная форма | Интегральная форма | |||
| 0-й | 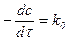
| 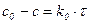
| 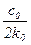
| моль/(дм3·с) |
| 1-й | 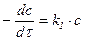
| 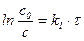
| 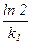
| с-1 |
| 2-й | 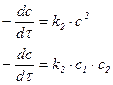
| 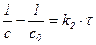
| 
| дм3/(моль·с) |
| 3-й | 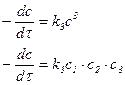
| 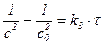
| 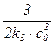
| дм3/(моль2·с) |
Примечание: с1, с2 … содержание разных исходных компонентов, в этом случае скорость определяется по каждому компоненту.
Время полупревращения является важной характеристикой химической реакции – это период времени, выраженный в единицах времени, за который прореагировала половина исходного вещества. Формулы для расчёта времени полупревращения легко выводятся из интегральной формы кинетического уравнения скорости.
Из уравнения Аррениуса видно, что зависимость скорости химической реакции (u @ k) от температуры имеет экспоненциальный характер, это согласуется с концепцией «активных частиц» и теорией «активных столкновений» в химической кинетике. Энергию активации в простейшем варианте можно определить, зная константы скорости реакции при двух разных температурах:
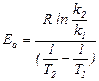 .
.
Зависимость скорости химической реакции от температуры в небольшом интервале температур описывает эмпирическое правило Вант-Гоффа: при изменении температуры на каждые 10 градусов скорость большинства химических реакций изменяется в 2–4 раза.
Математическое выражение правила Вант-Гоффа:
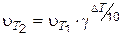 ,
,
где 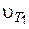 и
и 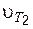 – скорости химических реакций при температурах Т 1 и Т 2;
– скорости химических реакций при температурах Т 1 и Т 2;
g – температурный коэффициент (коэффициент Вант-Гоффа), определяется экспериментально.
Связь между температурным коэффициентом и энергией активации выражается уравнением
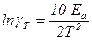 или
или  .
.
Теоретически кинетику реакций в газовой фазе описывает теория активных столкновений. В которой очевидна идея о том, что химическое превращение происходит при соприкосновении молекул, поскольку самопроизвольный распад молекул может быть только исключением. Предполагается, что скорость реакции равна числу соударений с энергией, не меньшей энергии активации. Тем самым, мы считаем, что протекание превращения не нарушает статистического равновесного распределения молекул по энергиям (распределения Максвелла - Больцмана). Это справедливо только в том случае, когда скорость превращения много меньше скорости перераспределения энергии. Сопоставление общего числа соударений и числа активных показывает, что между актами реакции, требующей энергии активации, происходит большое число неактивных соударений. Т.е. допущение о равновесии не кажется невероятным, поскольку перераспределение поступательной энергии происходит при каждом или нескольких ударах. Основные уравнения теории активных соударений были получены Траутцом, 1916 г, и Льюисом, 1918 г.
Очевидно, что скорость реакции (A + B → продукты) равна
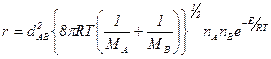 , для одинаковых молекул
, для одинаковых молекул
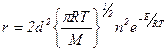
Тогда константы скорости выразятся как:
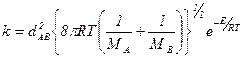
а для одинаковых молекул –
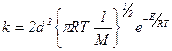
Окончательно, константа скорости
k = Zoe − E / RT ,
где Z o соответствует общему числу соударений при концентрациях, равных 1.
Дифференцирование основной формулы ТАС дает:
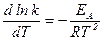
Разница невелика Е и ЕА: Е = ЕА + RT - при 500 К она примерно 4 кДж/моль, что обычно находится в пределах ошибки определения энергии активации в кинетических опытах, и часто этой разницей можно пренебречь. Основная заслуга ТАС состоит в том, что она позволила получить закон Аррениуса из молекулярной модели, дала физическое обоснование энергии активации и, в известной степени, предэкспоненциальному множителю. Теория позволяет в ряде случаев даже оценить величину константы скорости с хорошей точностью.
В 80-е годы XIX в. изучением скоростей химических реакций занимался В. Оствальд (Рига), сформулировавший закон разбавления (разведения), который является частным случаем закона действующих масс в применении к слабым электролитам:
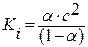 ,
,
где Ki –константа ионизации слабого электролита; a-степень диссоциации (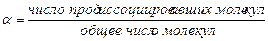 ); с – концентрация электролита.
); с – концентрация электролита.
Сравнение В.Оствальдом относительной активности различных кислот, привело его к выяснению условий химического равновесия и к развитию концепции катализа.
Катализ, как средство управления химическими процессами, был открыт в 1812 г. академиком Петербургской академии наук К.С. Кирхгофом (1764–1833). Катализаторы – вещества, ускоряющие реакцию, но не участвующие в стехиометрических продуктах реакции. Катализаторы влияют на энергетику реагирующих частиц, образуя с ними промежуточные соединения. Иногда катализирующим действием обладают продукты реакции, образование которых приводит к ускорению процесса. Такие реакции называются автокаталитическими.
Обычно химические реакции монотонно приближаются к устойчивому конечному состоянию.
Относительно редко, в ходе некоторых сложных реакций, концентрации промежуточных соединений и скорость реакции испытывают колебания. Колебательные реакции лежат в основе ряда важнейших биологических и природных процессов – генерации биоритмов, нервных импульсов, мышечного сокращения.
Химические колебательные реакции используют при создании химических лазеров.
Применение катализаторов коренным образом преобразовало всю химическую промышленность.
Современная каталитическая химия включает металлокомплексный катализ, межфазный катализ, мицеллярный катализ (посредством коллоидных систем), мембранный катализ (с участием веществ, действующих как молекулярные сита), катализ посредством ферментоподобных систем. В каталитической химии предприняты попытки моделировать природные катализаторы – ферменты, ускоряющие химические реакции в мягких условиях – при комнатной температуре и обычном давлении.
Созданы иммунобилизованные катализаторы – природные ферменты, привитые на твердую основу. И тем не менее, катализ во многом еще остается загадкой природы.
|
|