Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Изомерия
|
|
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии
Не следует путать с изомерией атомных ядер.
Изомерия (от др.-греч. ἴ σ ο ς — равный + μ έ ρ ο ς — доля, часть) — явление, заключающееся в существовании химических соединений — изомеров, — одинаковых по атомному составу и молекулярной массе, но различающихся по строению или расположению атомов в пространстве и, вследствие этого, по свойствам.
Содержание
[убрать]
· 1 Исторические сведения
· 2 Структурная изомерия
o 2.1 Изомерия углеродной цепи (углеродного скелета)
o 2.2 Валентная изомерия
o 2.3 Изомерия функциональной группы (межклассовая изомерия)
o 2.4 Изомерия положения
o 2.5 Метамерия
· 3 Пространственная изомерия (стереоизомерия)
o 3.1 Энантиомерия (оптическая изомерия)
o 3.2 Диастереомерия
§ 3.2.1 σ —диастереомерия
§ 3.2.2 π —диастереомерия (геометрическая изомерия)
· 4 Изомеризация
· 5 Литература
· 6 См. также
Исторические сведения[править | править вики-текст]
В итоге дискуссии Ю. Либиха и Ф. Вёлера в 1823 году было установлено, что существуют два резко различных по свойствам вещества состава AgCNO — циановокислое (AgNCO (англ.)русск.) и гремучее (AgONC) серебро. Ещё одним примером послужили винная и виноградная кислоты, после исследования которых Й. Берцелиус в 1830 году ввёл термин «изомерия» и высказал предположение, что различия возникают из-за «различного распределения простых атомов в сложном атоме» (то есть, в современных терминах, молекуле).
Подлинное объяснение изомерия получила лишь во 2-й половине XIX века на основе теории химического строения А. М. Бутлерова (структурная изомерия) и стереохимического учения Я. Г. Вант-Гоффа(пространственная изомерия).
Структурная изомерия[править | править вики-текст]
Структурная изомерия — результат различий в химическом строении. К этому типу относят:
Изомерия углеродной цепи (углеродного скелета)[править | править вики-текст]
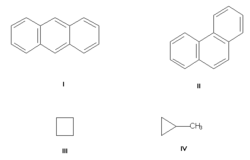
Изомерия углеродного скелета, обусловленная различным порядком связи атомов углерода. Простейший пример — бутан СН3—СН2—СН2—СН3 и изобутан(СН3)3СН. Другие примеры: антрацен и фенантрен (формулы I и II, соответственно), циклобутан и метилциклопропан (III и IV).
Валентная изомерия[править | править вики-текст]
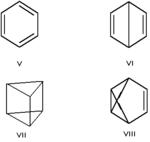
Валентная — особый вид структурной изомерии, при которой изомеры можно перевести друг в друга лишь за счёт перераспределения связей. Например, валентными изомерами бензола (V) являются бицикло[2.2.0]гекса-2, 5-диен (VI, «бензол Дьюара»), призман (VII, «бензол Ладенбурга»), бензвален (VIII).
Изомерия функциональной группы (межклассовая изомерия)[править | править вики-текст]
Различается характером функциональной группы; например, этанол (CH3—CH2—OH) и диметиловый эфир (CH3—O—CH3).
Изомерия положения[править | править вики-текст]
Тип структурной изомерии, характеризующийся различием положения одинаковых функциональных групп или кратных связей при одинаковом углеродном скелете. Пример: 2-хлорбутановая кислота и 4-хлорбутановая кислота.
Метамерия[править | править вики-текст]
Метамерия — вид структурной изомерии, для которого характерно различное распределение углеродных атомов между несколькими углеводородными радикалами, разделенными в молекулегетероатомом. Метамерия известна в рядах алифатических простых эфиров, сложных эфиров, тиоспиртов и аминов. В настоящее время термин используется редко.
На данный вид изомерии ещё указывал А. М. Бутлеров, называя его «изомерия нецельных структур».
Пример: CH3CH2OCH2CH3 — диэтиловый эфир и CH3OCH2CH2CH3 — метилпропиловый эфир
Пространственная изомерия (стереоизомерия)[править | править вики-текст]
Основная статья: Стереоизомеры
Пространственная изомерия (стереоизомерия) возникает в результате различий в пространственной конфигурации молекул, имеющих одинаковое химическое строение. Для обозначения пространственных изомеров разных типов разработана стереохимическая номенклатура, собранная в разделе E номенклатурных правил ИЮПАК по химии[ источник не указан 1591 день ].
Этот тип изомерии подразделяют на энантиомерию (оптическую изомерию) и диастереомерию.
Энантиомерия (оптическая изомерия)[править | править вики-текст]
Основная статья: Оптическая изомерия
Энантиомерами (оптическими изомерами, зеркальными изомерами) являются пары оптических антиподов — веществ, характеризующихся противоположными по знаку и одинаковыми по величине вращениями плоскости поляризации света при идентичности всех других физических и химических свойств (за исключением реакций с другими оптически активными веществами и физических свойств в хиральной среде). Необходимая и достаточная причина возникновения оптических антиподов — принадлежность молекулы к одной из следующих точечных групп симметрии: C n, D n, T, O или I (хиральность). Чаще всего речь идет об асимметрическом атоме углерода, то есть об атоме, связанном с четырьмя разными заместителями.
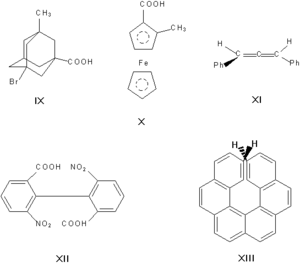
Асимметрическими могут быть и другие атомы, например атомы кремния, азота, фосфора, серы. Наличие асимметрического атома — не единственная причина энантиомерии. Так, имеют оптические антиподы производные адамантана (IX), ферроцена (X), 1, 3-дифенилаллена (XI), 6, 6'-динитро-2, 2'-дифеновой кислоты (XII). Причина оптической активности последнего соединения — атропоизомерия, то есть пространственная изомерия, вызванная отсутствием вращения вокруг простой связи. Энантиомерия также проявляется в спиральных конформациях белков, нуклеиновых кислот, в гексагелицене(XIII).
Диастереомерия[править | править вики-текст]
Диастереомерными считают любые комбинации пространственных изомеров, не составляющие пару оптических антиподов. Различают σ - и π -диастереомеры.
σ —диастереомерия[править | править вики-текст]
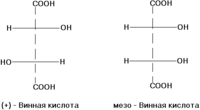
σ -диастереомеры отличаются друг от друга конфигурацией части имеющихся в них элементов хиральности. Так, диастереомерами являются (+)-винная кислота и мезо-винная кислота, D-глюкоза и D-манноза, например:
π —диастереомерия (геометрическая изомерия)[править | править вики-текст]
π -диастереомеры, называемые также геометрическими изомерами, отличаются друг от друга различным пространственным расположением заместителей относительно плоскости двойной связи (чаще всего С=С и С=N) или цикла. К ним относятся, например, малеиновая и фумаровая кислоты (формулы XIV и XV соответственно), (Е)- и (Z)-бензальдоксимы (XVI и XVII), цис- и транс-1, 2-диметилциклопентаны (XVIII и XIX).
Изомеризация[править | править вики-текст]
Химические превращения, в результате которых структурные изомеры превращаются друг в друга, называется изомеризацией. Такие процессы имеют важное значение в промышленности. Так, например, проводят изомеризацию нормальных алканов в изоалканы для повышения октанового числа моторных топлив; изомеризуют пентан в изопентан для последующего дегидрирования в изопрен. Изомеризацией являются и внутримолекулярные перегруппировки, из которых большое значение имеет, например, перегруппировка Бекмана — превращение циклогексаноноксима в капролактам(сырьё для производства капрона).
Процесс взаимопревращения энантиомеров называется рацемизацией: она приводит к исчезновению оптической активности в результате образования эквимолярной смеси (−)- и (+)-форм, то есть рацемата. Взаимопревращение диастереомеров приводит к образованию смеси, в которой преобладает термодинамически более устойчивая форма. В случае π -диастереомеров это обычно транс-форма. Взаимопревращение конформационных изомеров называется конформационным равновесием.
Явление изомерии в огромной степени способствует росту числа известных (и ещё в большей степени — числа потенциально возможных) соединений. Так, возможное число структурно-изомерных дециловых спиртов — более 500 (известно из них около 70), пространственных изомеров здесь более 1500.
При теоретическом рассмотрении проблем изомерии все большее распространение получают топологические методы; для подсчёта числа изомеров выведены математические формулы.
Структурная химия
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии
Структурная химия — раздел, область химии, изучающая связь различных физических и физико-химических свойств различных веществ с их химическим строением и реакционной способностью. Структурная химия рассматривает не только геометрическое строение молекул; изучению подвергается следующее — длины химических связей, валентные углы, координационные числа, конформациии конфигурации молекул; эффекты их взаимного влияния, ароматичность.
Содержание
[убрать]
· 1 Способы изучения
· 2 История развития науки
· 3 См. также
· 4 Литература
· 5 Ссылки
Способы изучения[править | править вики-текст]
Структурная химия базируется на следующих экспериментальных способах изучения веществ:
· рентгеновский структурный анализ,
· инфракрасная спектроскопия,
· ультрафиолетовая спектроскопия,
· фотоэлектронная спектроскопия,
· нейтронография,
· электронография,
· спектроскопия комбинационного рассеивания,
· микроволновая спектроскопия,
· резонансные методы:
· ЯМР, ЭПР, мёссбауэровская спектроскопия, ядерный квадрупольный резонанс
· адсорбция,
· катализ и др.
История развития науки[править | править вики-текст]
В 1857-м году, ученый Кекуле, исходя из теории валентности (под валентностью имелось в виду количество атомов водорода, соединенных с одним атомом элемента), смог предположить, что углеродчетырёхвалентен, исходя из этого он может соединиться с четырьмя другими атомами, образуя длинные цепи — прямые или разветвленные. Поэтому органические молекулы стали изображать не в виде комбинаций радикалов, а в виде структурных формул атомов и связей между ними. К 1860-му году трудами ученых Кекуле и Бутлерова была заложена основа структурной химии, которая позволяла объяснять свойства веществ, исходя из расположения атомов в их молекулах. Впоследствии, в 1874-м году, датский химик Якоб Вант-Гофф и французский химик Жозеф Ашиль Ле Бель распространили идею о расположении атомов в пространстве. Они считали, что молекулы представляют собой не плоские, а объемные структуры. Эта концепция позволяла объяснить многие известные явления, например пространственную изомерию, существование молекул одинакового состава, но с разными свойствами. Очень хорошо вписывались в неё данные Луи Пастера о изомерах винной кислоты.
К концу XIX века идеи структурной химии были подкреплены данными, полученными спектроскопическими методами. Эти методы позволяли получать информацию о строении молекул исходя из ихспектров поглощения. К началу 20-го века концепция объемной организации молекул сложных органических и неорганических соединений была принята практически всеми учеными.
Электронная теория химической связи
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии
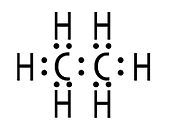
Рис.1.
Электронная теория химической связи была предложена и развита американским физикохимиком Льюисом Г.Н в 1912—1916 гг[1]. Ковалентная химическая связь, по Льюису, образуется за счёт обобществления пары электронов, то есть электронная плотность распределяется между двумя атомами, в противовес господствующей в то время теории, будто один из связанных атомов несёт положительный, а другой отрицательный заряд. Льюис также предложил обозначать электроны точками у символахимического элемента. Электронная теория химической связи включает идею Льюиса, что завершённый внешний электронный слой атома содержит восемь электронов.
Электронная теория химической связи Льюиса стала основой классической теории строения в органической химии, базирующейся на представлении о парной связи между атомами, образованной дублетом электронов. Например, связь C-C в молекуле этана изображается как пара электронов, принадлежащая обоим атомам углерода (рис.1).
При этом электроны атомов водорода дозаполняют электронные оболочки обоих атомов углерода до октета (правило октета).
Образование химической связи может происходить как путём обобществления двух электронов, принадлежащих разным атомам или свободным радикалам
R· + R· → R: R,
так и в результате передачи пары электронов одним из атомов в общее пользование с другим, электронодефицитным атомом по донорно-акцепторному механизму:
X+ +: Y- → X: Y
Концепции электронной теории химической связи — общепринятый язык теоретической органической химии[2].
Образование ковалентной связи, осуществляемое, по Льюису, общей для двух атомов электронной парой (дублетом электронов) впоследствии было интерпретировано в рамках квантовой механикиВ.Гайтлером и Ф.Лондоном как эффект перекрывания электронных плотностей (атомных орбиталей) взаимодействующих атомов. В рамках этой теории (теория валентных схем) образование валентной связи между двумя атомами обусловлено взаимной компенсацией спинов их валентных электронов, причём получающаяся электронная пара входит во внешние электронные оболочки обоих атомов.
Концепции электронной теории химической связи и её квантовомеханического дополнения — теории валентных схем оказалась не применимыми для описания структуры «неклассического» иона —молекулярного иона водорода H2+ — молекулярного соединения, образованного единственным электроном. В молекулярном ионе водорода H2+ нет ни дублета электронов, нет ни компенсации спинов электронов, нет ни перекрывания атомных орбиталей, которые образуют ковалентную связь, поскольку в системе присутствует лишь единственный электрон. Тем не менее, молекулярный ион водорода H2+ является устойчивым соединением[3].
Содержание
[убрать]
· 1 Эволюция представления об электронной паре
· 2 Строение связывающей электронной пары
· 3 Примечания
· 4 См. также
Эволюция представления об электронной паре[править | править вики-текст]
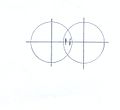
Рис.1.Спин-спиновое взаимодействие электронов в связывающей электронной паре
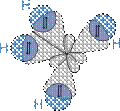
Рис.2.Модель молекулы метана, образованной sp3-гибридными орбиталями
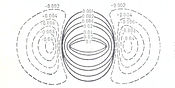
Рис.3.Контурная карта электронной плотности для молекулы водорода
Связывающую электронную пару первоначально представляли двумя точками, расположенными между двумя атомами молекулы.
С появлением теории валентных схем и открытием спина электрона, электронную пару стали представлять двумя параллельными стрелками, направленными в противоположные стороны и расположенные в области перекрытия атомных орбиталей (рис.1)
В рамках теории валентных схем атом углерода в алканах, например, в метане, окружался четырьмя связывающими электронными парами (рис.2)
В теории молекулярных орбиталей связывающую электронную пару стали представлять в форме контурных карт электронной плотности. На рис.3 представлена контурная карта разностей электронной плотности для молекулы водорода (сплошные линии — области увеличения электронной плотности (в единицах заряда электрона)), пунктирные — области её уменьшения по сравнению с электронной плотностью несвязанных атомов водорода, сближенных на равновесное расстояние[4].
Во всех случаях связывающая электронная пара или электронная плотность концентрируется на линии, соединяющей ядра молекулы. В теории отталкивания валентных электронных пар проведена оценка размеров электронных облаков связывающих электронных пар, Å [5]:
| Второй период | Третий период | Четвёртый период | ||||
| элемент | re | элемент | re | элемент | re | |
| Li | 0, 86 | Na | 0, 85 | K | 0, 87 | |
| Be | 0, 74 | Mg | 0, 85 | Ca | 0, 81 | |
| B | 0, 61 | Al | 0, 75 | Ga | 0, 68 | |
| C | 0, 62 | Si | 0, 76 | Ge | 0, 69 | |
| N | 0, 59 | P | 0, 76 | As | 0, 74 | |
| O | 0, 57 | S | 0, 75 | Se | 0, 75 | |
| F | 0, 57 | Cl | 0, 73 | Br | 0, 75 | |
| Ne | 0, 56 | Ar | 0, 72 | Kr | 0, 75 |
Строение связывающей электронной пары[править | править вики-текст]
Для описания химических соединений с кратными связями Л.Полинг вводит представления о σ - и π -связывающих электронных парах. Электронная пара, образующая σ -связь, обладает, по Полингу, цилиндрической симметрией относительно линии связи, в то время как, электронная пара, образующая π -связь, антисимметрична относительно плоскости, проходящей через связь. Предполагалось, что двойная связь состоит из одной σ -и одной π -связи, а тройная — из одной σ - и двух ортогональных π -связей.[6]
В такой интерпретации связывающие электронные пары становились неравноценными — σ -связи располагались по линии, соединяющей ядра молекулы, а электронные пары π -связи вытеснялись с этой линии (рис.4.)
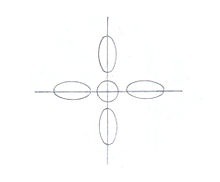
Рис.4
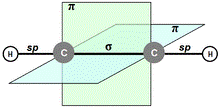
Рис.4.Тройная связь в молекуле ацетилена и осевая проекция
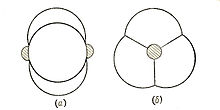
Рис.5
Позднее, на симпозиуме, посвящённом памяти Кекуле (Лондон, сентябрь 1958 г.), Л.Полинг отказался от σ -, π - описания кратных связей и предложил теорию изогнутой химической связи, в которой все связывающие электронные пары равноценны и удалены от линии, соединяющей ядра атомов молекулы. Двойная связь представляется как комбинация двух, а тройная — трёх одинаковых изогнутых химических связей.[7]
Зная межъядерные расстояния ординарной химической связи (1, 54 Å), двойной химической связи (1, 33 Å) и тройной химической связи (1, 20 Å)[7] можно было вычислить величины прогиба ординарной химической связи как в двойной связи (0, 388 Å), так и в тройной связи (0, 483 Å).
В теории отталкивания электронных пар три связывающие электронные пары тройной связи представлены в виде трёх сплющенных сфер, дающих форму диска или сплющенного эллипсоида, расположенных вокруг линии, соединяющей ядра атомов молекулы ацетилена (рис.5).
Теория изогнутой химической связи и теория отталкивания электронных пар, в отличие от теории валентных связей и теории молекулярных орбиталей, учитывали электростатическое отталкивание электронов (кулоновскую электронную корреляцию).
Электростатическое отталкивание электронов непосредственно в связывающей электронной паре было заложено в боровской химической связи. Нильс Бор представлял электронную пару в форму кольца электронов. В метане, по Бору:
Ядро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами [8]
В настоящее время дают следующее трактовку связывающей электронной пары (рис.6):
Простую модель двухатомной связи можно представить в виде двух ядер, между которыми в плоскости, перпендикулярной оси, соединяющей ядра, вращаются связывающие электроны. При этом предполагается, что между заряженными частицами (электроны, ядра) действуют только электростатические силы. Если соединяющиеся в молекулу атомы имеют одинаковые потенциалы ионизации, плоскость вращения электронов располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны, то плоскость вращения электронной пары смещается в сторону атома с большим ППИ. [9]
(ППИ — первый потенциал ионизации)
Дисперсионные силы
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии
Дисперсионные силы (дисперсионное притяжение, Лондоновские силы, Лондоновские дисперсионные силы, LDF) — силы электростатического притяжения мгновенного и индуцированного (наведённого) диполей электрически нейтральных атомов или молекул.
Происхождение дисперсионных сил было объяснено в 1930 году Фрицем Лондоном — немецким физиком-теоретиком[1]. Дисперсионные силы универсальны (то есть проявляются во всех случаях), так как они обусловлены взаимодействием атомов и молекул друг с другом за счёт их дипольных моментов, собственных или взаимоиндуцированных. Считается, что дисперсионная энергия не имеет классического аналога и определяется квантовомеханическими флуктуациями электронной плотности. Мгновенное распределение заряда одного атома или молекулы, характеризуемое мгновенным дипольным моментом, индуцирует мгновенный дипольный момент в другом атоме или молекуле[2]. При сближении атомов или молекул ориентация микродиполей перестаёт быть независимой, и их появление и исчезновение в разных атомах и молекулах происходит в такт друг к другу. Синхронное появление и исчезновение микродиполей разных атомов и молекул сопровождается их притяжением.[3]
В результате возникает взаимодействие этих моментов. Потенциальная энергия дисперсионного взаимодействия Eдисп. = -C/R6. Коэффициент C приближённо вычисляют по формуле:
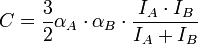 , где
, где 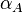 и
и 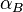 — электронные поляризуемости атомов или молекул,
— электронные поляризуемости атомов или молекул, 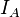 и
и 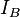 — потенциалы ионизации атомов или молекул, R — расстояние между атомами и молекулами.[2]
— потенциалы ионизации атомов или молекул, R — расстояние между атомами и молекулами.[2]
Дисперсионное взаимодействие (притяжение) возникает между всеми атомами и молекулами.
Межатомное взаимодействие сопровождается рекомбинацией атомов с образованием молекулы за исключением атомов инертных газов, которые сохраняют свою индивидуальность. Так, все инертные газы при нормальных условиях моноатомны. Однако, дисперсионные силы обуславливают возможность существования различных агрегатных состояний инертных газов (газ, жидкость и твёрдые тела).
Теория отталкивания электронных пар
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии
Теория отталкивания электронных пар валентных орбиталей (ОЭПВО) — один из подходов в химии, необходимый для объяснения и предсказания геометрии молекул. Согласно этой теориимолекула всегда будет принимать форму, при которой отталкивание внешних электронных пар минимально (принцип минимума энергии).
Содержание
[убрать]
· 1 История
· 2 Описание
· 3 Развитие теории ОЭПВО и примеры
· 4 Недостатки теории ОЭПВО и отклонения от ее предсказаний
· 5 Размеры электронных облаков электронных пар
· 6 Рекомендуемая литература
· 7 Примечания
· 8 См. также
История[править | править вики-текст]
В 1940 г. Н. Сиджвик и Г. Пауэлл предложили модель отталкивания электронных пар, которая впоследствии была развита (1957) Р. Гиллеспи и Р. Найхолмом. Основные идеи этого подхода, приложимого только к соединениям непереходных элементов, сводятся к следующему:
1. Конфигурация связей многовалентного атома (или иона) обуславливается исключительно числом связывающих и несвязывающих электронных пар в валентной оболочке центрального атома.
2. Ориентация облаков электронных пар валентных орбиталей определяется максимальным взаимным отталкиванием заполняющих их электронов.
Описание[править | править вики-текст]
Если бы природа сил взаимного отталкивания электронных пар имела чисто электростатический характер, эти силы определялись бы соотношением (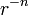 ), где
), где 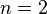 . Однако кроме электростатического взаимодействия электронные пары на разных локализованных молекулярных орбиталях (ЛМО) испытывают отталкивание еще в силу действия принципа Паули, поэтому в выражении для сил
. Однако кроме электростатического взаимодействия электронные пары на разных локализованных молекулярных орбиталях (ЛМО) испытывают отталкивание еще в силу действия принципа Паули, поэтому в выражении для сил 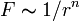
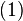 , где
, где  — расстояние между «центрами тяжести» облаков электронных пар ЛМО,
— расстояние между «центрами тяжести» облаков электронных пар ЛМО,  . Задача поиска расположения центров облаков электронных пар, расталкивающихся в соответствии с при равных для всех пар величинах , эквивалентна задаче размещения нескольких частиц на поверхности сферы при их максимальном удалении друг от друга. Эта задача решается строго для числа частиц от 2 до 12 и дает следующий результат:
. Задача поиска расположения центров облаков электронных пар, расталкивающихся в соответствии с при равных для всех пар величинах , эквивалентна задаче размещения нескольких частиц на поверхности сферы при их максимальном удалении друг от друга. Эта задача решается строго для числа частиц от 2 до 12 и дает следующий результат:
Таблица 1. Конфигурация связей центрального атома А в зависимости от числа электронных пар q на его валентных орбиталях.
| q | Конфигурация |
| Линейная | |
| Равносторонний треугольник | |
| Тетраэдр | |
| Тригональная бипирамида | |
| Октаэдр | |
| Октаэдр с дополнительной вершиной | |
| Квадратная антипризма | |
| Треугольная призма с тремя дополнительными вершинами | |
| Квадратная антипризма с двумя дополнительными вершинами | |
| Икосаэдр без одной вершины | |
| Икосаэдр |
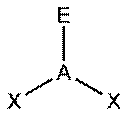
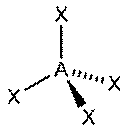
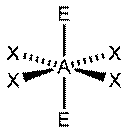
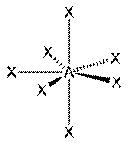
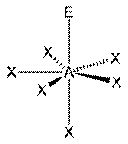
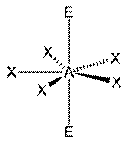
В таблице 1 в число q входят электронные пары как на связывающих ЛМО, так и на несвязывающих, то есть неподеленные электронные пары. Гиллеспи ввел для связывающих электронных пар обозначение X, а для несвязывающих — Е. С учетом этих обозначений можно следующим образом представить геометрическую конфигурацию молекул типа AXmEn. Как видно из данных таблицы 2 (приведены некоторые примеры), в рамках теории ОЭПВО для определения топологии связей центрального атома в молекулах, образованных непереходными элементами, необходимо только сосчитать число электронных пар на связывающих и несвязывающих орбиталях и разместить их на осях соответствующего многогранника.
Таблица 2-3. Геометрия структуры молекул типа AXmEn без кратных связей.
| Общее число электронных пар. | Геометрия 0 свободных пар | 1 свободная пара | 2 свободные пары | 3 свободные пары |
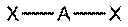 Линейная
Линейная
| ||||
 Равносторонний треугольник
Равносторонний треугольник
|  Искаженная
Искаженная
| |||
 Тетраэдр
Тетраэдр
| 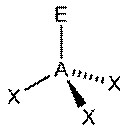 Тригональная пирамида
Тригональная пирамида
| 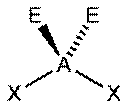 Искаженная
Искаженная
| ||
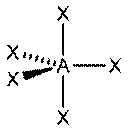 Тригональная бипирамида
Тригональная бипирамида
| 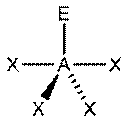 Бисфеноид
Бисфеноид
| 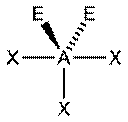 Т-форма
Т-форма
| 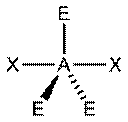 Искажённая
Искажённая
| |
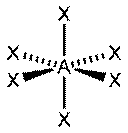 Октаэдр
Октаэдр
| 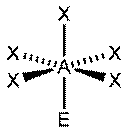 Квадратная пирамида
Квадратная пирамида
|  Плоский квадрат
Плоский квадрат
| ||
 Пентагональная бипирамида
Пентагональная бипирамида
|  Пентагональная пирамида
Пентагональная пирамида
| 
|
| Тип молекулы | Конфигурация | Расположение электронных пар† | Геометрия‡ | Примеры |
| AX1En | Двухатомная | 
|
| HF, O2 |
| AX2E0 | Линейная | 
| 
| BeCl2, HgCl2, CO2 |
| AX2E1 | Искаженная | 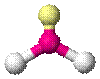
| 
| NO2− , SO2, O3 |
| AX2E2 | Искаженная | 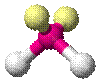
|
| H2O, OF2 |
| AX2E3 | Линейная | 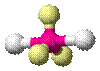
|
| XeF2, I3− |
| AX3E0 | Равносторонний треугольник | 
| 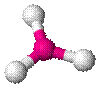
| BF3, CO32− , NO3− , SO3 |
| AX3E1 | Тригональная пирамида | 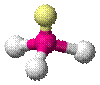
| 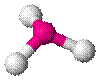
| NH3, PCl3 |
| AX3E2 | Т-образная | 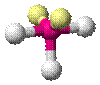
| 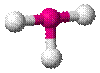
| ClF3, BrF3 |
| AX4E0 | Тетраэдр | 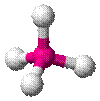
| 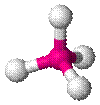
| CH4, PO43− , SO42− , ClO4− |
| AX4E1 | Дисфеноид («Качели») | 
| 
| SF4 |
| AX4E2 | Плоскоквадратная геометрия | 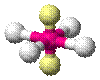
| 
| XeF4 |
| AX5E0 | Тригональная бипирамида | 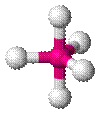
|
| PCl5 |
| AX5E1 | Квадратная пирамида | 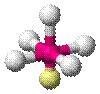
| 
| ClF5, BrF5 |
| AX5E2 | 
| 
| XeF5- | |
| AX6E0 | Октаэдр | 
| 
| SF6 |
| AX6E1 | Пентагональная пирамида | 
| 
| XeF6 |
| AX7E0 | Пентагональная бипирамида | 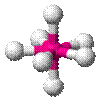
| 
| IF7 |
† Расположение электронных пар, включая свободные (помечены желтым цветом)
‡ Геометрия (без свободных элетронных пар)
Развитие теории ОЭПВО и примеры[править | править вики-текст]
1. Неподеленная электронная пара занимает больший объем, чем пара электронов на орбитали, участвующей в образовании ординарной связи. Сила отталкивания электронных пар в данной валентной оболочке понижается в следующем порядке: неподеленная пара — неподеленная пара (Е — Е) > неподеленная пара — связывающая пара (Е—X) > связывающая пара — связывающая пара (X—X).
Это допущение вытекает из таких простых аргументов, как то, что неподеленная электронная пара находится в поле только одного положительного атомного остова и, следовательно, более диффузна, чем связывающая электронная пара, испытывающая сжатие полем зарядов двух атомных остовов. Отсюда следует вывод, что в серии изоэлектронных молекул (то есть содержащих равное число электронных пар в валентной оболочке) неподеленная пара, заменяя связывающую электронную пару, стремится занять больше пространства вокруг атома. Это уменьшает валентные углы между связями, например, в ряду
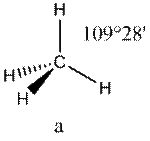
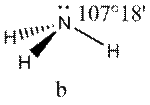
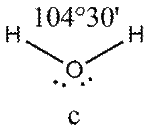
Аналогичное объяснение имеют отклонения от полностью симметричной ориентации связей в молекуле типа АХ5Е, АХ4Е, АХ3Е2.
Поскольку отталкивания электронных пар типа Е — X доминируют по сравнению с отталкиваниями X—X, валентные углы ХАХ несколько сжаты:
:

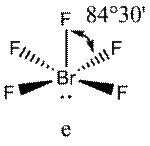
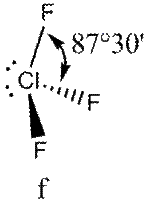
В прямой связи с данными об эффективных объемах связывающих и неподеленных электронных пар находится такое важное следствие, как положение о том, что в молекулах типа АХ4Е, АХ3Е2, АХ2Е3, геометрическая форма которых производится от структуры тригональной бипирамиды, электронные пары всегда занимают экваториальные положения (см. Таблица 2-3)
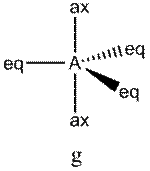
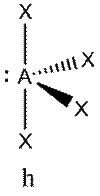
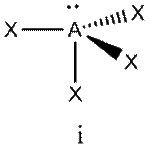
Это объясняется тем, что в структуре h имеются только два невыгодных Е—Х-взаимодействия электронных пар, когда их оси составляют угол 90°. В структуре i (топомерной h) таких невыгодных взаимодействий три.
2. Объем электронной пары, участвующей в образовании связи, уменьшается с увеличением электроотрицательности лиганда.
Более электроотрицательный лиганд сильнее притягивает общее электронное облако связывающей пары, что можно представить как дополнительное сжатие этого облака. Данная электронная пара будет более удалена от центрального атома и испытывает меньшее отталкивание со стороны других соседних электронных пар. Все это поведет к тому, что валентные углы, составляемые связями центрального атома с наиболее электроотрицательными лигандами, должны иметь наименьшие значения.
Эффект влияния изменения электроотрицательности лигандов на валентные углы связей иллюстрируется сравнением молекул NH3 и NF3. Большая электроотрицательность фтора уменьшает размеры пары на связи N—F, в результате углы FNF составляют всего 102°, что на 5° меньше, чем углы HNH в аммиаке. Такая же тенденция наблюдается в ряду РI3 (102°), РВr3 (101, 5°), РСl3(100, 3°), РF3(97, 8°).
Интересный пример — молекула (СН3)2РF3.
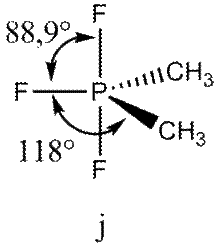
Валентные углы в этой молекуле уменьшаются в порядке СРС > СРF > FРF, соответствующем порядку возрастания электроотрицательности лигандов.
Следует отметить, что, если рассматривать неподеленную электронную пару как некий воображаемый лиганд (фантом-лиганд) с предельно малой электроотрицательностью, правила 1 и 2 легко обобщаются.
3. Две электронные пары двойной связи или три электронные пары тройной связи занимают больший объем, чем электронная пара одинарной связи.
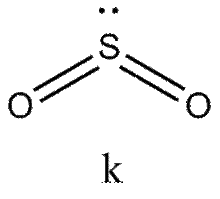
Это правило является основным при рассмотрении геометрической структуры молекул, содержащих кратные связи. Как и для остальных молекул, определение конфигурации связей центрального атома основывается на выделении электронных пар σ -связей и неподеленных электронов. Электроны π -связей на этой стадии не учитываются. Так, например, чтобы определить форму молекулы S02 (k), необходимо учесть, что из шести электронов в валентной оболочке серы два расходуются на образование двух π -связей. Из оставшихся четырех два образуют с неподеленными электронами кислородных атомов а-связи, а два остаются в виде неподеленной пары. Таким образом, необходимо учесть относительную ориентацию облаков трех электронных пар, что в согласии с данными табл. 4 ведет к угловой конфигурации.
В табл. 5 представлены данные о геометрической структуре широкого ряда молекул непереходных элементов с кратными связями. Так как кратная связь содержит более чем одну электронную пару, ее электронное облако занимает большее пространство, чем электронная пара ординарной связи. Размер электронного облака двойной связи по сравнению с размером орбитали неподеленной электронной пары недостаточно определен. Обычно их размеры принимают равными. Больший размер кратной связи виден из примеров молекул типа Х2СО и Х2С=СН2, пирамидальных молекул типа Х2SO, тетраэдрических молекул типа РОХ3. Из табл. 5 видно, что угол ХСХ всегда меньше 120°, угол ХS0 больше угла ХSХ.
Таблица 4. Геометрическая структура молекул непереходных элементов, содержащих кратные связи.
| Общее число электронных пар σ -орбиталей и несвязывающих орбиталей | Число σ -связей | Число неподеленных пар | Конфигурация связей | Примеры |
| Линейная | 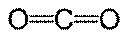 , , 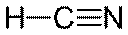
| |||
| Треугольная Угловая | 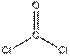 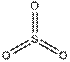 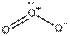 , , 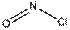
| |||
| Тетраэдрическая Пирамидальная Угловая | 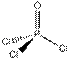 , , 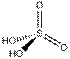  
| |||
| Тригональная бипирамида Бисфеноидная | 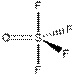 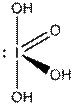
| |||
| Октаэдрическая | 
|
Важно подчеркнуть, что выводы теории ОЭПВО о геометрическом строении молекул легко экстраполируются на более сложные молекулы и ионы, чем рассмотренные в табл. 2-5. В каждом случае необходимо выделить фрагмент, содержащий центральный атом, координирующий около себя другие атомы или их группировки, и установить число и тип окружающих данный атом электронных пар. Таким образом, нетрудно определить, например, структуру молекулы Р4. Каждый атом фосфора в ней имеет три соседа и, кроме того, сохраняет одну неподеленную электронную пару. Следовательно, должна реализоваться тетраэдрическая конфигурация осей электронных пар, отвечающая молекулярной структуре l:

| Молекула | Угол, град | Молекула | Угол, град | ||
| XCX | CXO | XCX | XCC | ||
| F2CO | 108, 0 | H2CH=CH2 | 116, 8 | ||
| CH3COF | 128; 122 | H2C=CHF | 115, 4 | 123, 3; 120, 9 | |
| Cl2CO | 111, 3 | 124, 3 | H2C=CF2 | 109, 3 | 125, 3 |
| H2CO | 115, 8 | 122, 1 | H2C=CCl2 | ||
| (NH2)2CO | F2C=CH2 | ||||
| (NH2)2CS | F2C=CFCl | ||||
| Молекула | XSX | XSO | Молекула | XPX | XPO |
| F2SO | 92, 8 | 106, 8 | POF3 | 101, 3 | |
| Br2SO | POCl3 | 103, 3 | |||
| (CH3)2SO | POBr3 | ||||
| (C6H5)2SO | 97, 3 | 106, 2 | PSF3 | 100, 3 | 113, 8 |
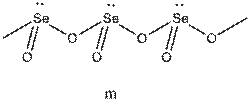
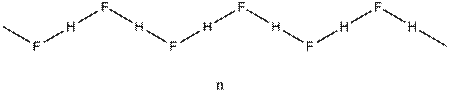
Таким же образом приходим к пирамидальной конфигурации связей атома селена (m) в полимерном диоксиде селена (SeO2)n, угловой структуре полимерной цепи кристалла НF (n). Положения теории ОЭПВО полезны при определении строения не только стабильных молекул и ионов, но также для описания относительной ориентации групп в метастабильных комплексах и даже переходных состояниях реакций присоединения, замещения. Так, например, в полном согласии с данными строгих расчетов предсказывается тригонально-бипирамидальное строение переходного состояния в реакции бимолекулярного замещения на sp3-углеродном центре:
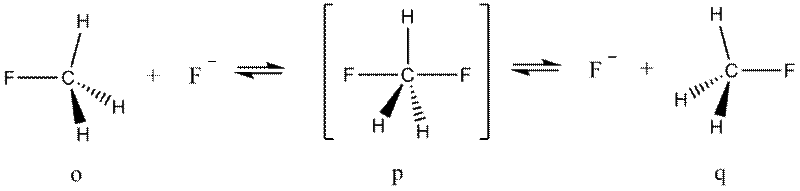
Действительно, пять электронных пар, окружающих центральный атом углерода в переходном состоянии реакции замещения, обусловливает его структуру p.
Недостатки теории ОЭПВО и отклонения от ее предсказаний[править | править вики-текст]
Как и всякая приближенная теория, основанная на той или иной модели, теория ОЭПВО сталкивается с рядом трудностей, предопределенных недостатками модели, лежащей в ее основе. Укажем на некоторые из них.
1 Как было отмечено в предыдущих разделах, теория приложима к описанию строения молекул только непереходных элементов, то есть элементов, не имеющих не полностью заполненные внутренние электронные оболочки. Дело в том, что наличие таких оболочек, например d -электронов в атомах переходных элементов, приводит к отклонениям от сферической симметрии распределения электронов остова. Это, в свою очередь, ведет к тому, что распределение облаков электронных пар в пространстве относительно центрального атома не подчиняется точно соотношению (1). Эти отклонения особенно заметны при значительном количестве (6—9) электронов в d -оболочках переходных элементов.
2 Участие d -орбиталей в связях, образуемых элементами низших периодов, также приводит к отклонениям от ожидаемой на основании представлений теории ОЭПВО геометрии. Хорошо известным примером являются угловые искажения молекул галогенидов щелочноземельных металлов. Эти отклонения иллюстрируются в табл. 6.
Таблица 6. Конфигурация связей в молекулах галогенидов щелочноземельных металлов МХ2 (л — линейная, у — угловая конфигурации)
| F | Cl | Br | I | |
| Be | л | л | л | л |
| Mg | у | л | л | л |
| Ca | у | л | л | л |
| Sr | у | у | л | л |
| Ba | у | у | у | у |
Причины этих отклонений вызваны изменениями в типе орбиталей центрального атома, образующих связи с галогенами, переходом от sp -типа к sd -типу по мере возрастания порядкового номера элемента и электроотрицательности лиганда. Теория ОЭПВО в отличие от представлений ЛМО и теории гибридизации АО не учитывает прямо тип орбиталей электронных пар, что и не позволяет учесть отдельные тонкие различия.
3 В соединениях типа АХ6Е и других с высоким координационным числом центрального атома неподеленная электронная пара является стереохимически инертной и структура соответствует конфигурации, получаемой без учета электронной пары Е. Так, анионы SbCl63-, ТеСl62- имеют октаэдрическое строение, хотя они, как и гексафторид ксенона ХеF6, содержат в валентной оболочке по семь электронных пар. Однако ХеF6 имеет в согласии с теорией ОЭПВО структуру неправильного октаэдра (табл. 3), тогда как в указанных анионах все связи равноценны. Другой пример — Сs2[XeF8]2-Анион этой соли, в котором центральный атом окружен девятью электронными парами, вопреки ожиданиям теории имеет строение квадратной антипризмы. Причина отмеченных отклонений состоит в том, что одна из валентных электронных пар, а именно ns2, сильно локализована и по своим свойствам резко отличается от характеристик остальных электронных пар.
4 Большие расхождения с предсказаниями теории ОЭПВО наблюдаются для соединений с высокополярными связями, близкими к ионному типу. Так, молекула Li2O, относящаяся к типу АХ2Е2, имеет не угловую, а линейную форму. Последнее понятно из электростатических соображений, если представить Li2O в форме ионной структуры Li+02-Li+.
5 В теории ОЭПВО характеристики заместителей X фактически не принимаются во внимание. Кроме неправильных предсказаний для ионных соединений, это ведет к неточному предсказанию и для соединений, в которых X представляет собой π -сопряженную систему. Так, анионы АХ3Е типа C(CN)3-, C(NO2)3- имеют не ожидаемую пирамидальную, а плоскую форму вследствие того, что последняя обеспечивает лучшие условия для включения неподеленной электронной пары в общую π -систему. Несмотря на отмеченные недостатки, представления теории ОЭПВО исключительно полезны и при правильном применении достаточно надежны для объяснения и предсказания структурных характеристик молекул и ионов, образованных непереходными элементами в самых различных валентных состояниях. Теория ОЭПВО может служить примером простой и эффективной теоретической концепции, позволяющей предвидеть главные детали молекулярной структуры без проведения трудоемких расчетов.
Размеры электронных облаков электронных пар[править | править вики-текст]
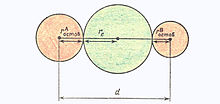
Рис. Строение ковалентной связи в модели жёстких сфер
В теории отталкивания электронных пар важное значение приобретают сведения об объёме электронных пар. Приближённая оценка объёмов электронных пар осуществляется с использованием модели жёстких сфер[1]. Согласно этой модели, длина ковалентной связи d равна сумме радиусов двух атомных остовов и диаметра связывающей электронной пары:
d = rAостов + rBостов + 2re,
Где re - радиус связывающей электронной пары (рис)
Для гомоядерный двухатомной молекулы ковалентный радиус атома rков = ½ d, поэтому справедливо следующее соотношение [1]:
rков = rостов + re или re = rков - rостов
Из этого соотношения принято рассчитывать радиусы электронных пар большинства элементов, используя значения ковалентных радиусов и ионных радиусов, которые соответствуют размерам атомных остовов.[1]
|
|