Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Коферменты, химическое строение и функции
|
|
Коферменты удобно классифицировать по структурно-физиологическим признакам и функциональным (каталитическим) свойствам. Структурно-физиологическая классификация одновременно учитывает происхождение и химическое строение коферментов:
Кофер менты
I. Витаминные ноферменты
1 Тиаминовые (ТМФ, ТДФ, ТГФ)
2 ' Флавиновые (ФМН. ФАД)
3 Пантотеновые (КоА, дефосфо КоА. 4-$ фогта нтотенат)
4 Никотннамидные (НАД. НАДФ) 5. Пиридоксиновые (ПАЛФ, ПАМФ) Ь. Фолиевые, или птеридиновые (ТГФК) 7. Кобамидные (метил коб ал а ми
д ен оз ил к о бала ми ч *
5 Биотиновые (карбоксибиотин) () Л иное вые (восстановленный ный липоамид)
10. Хиноновые (убихииои, пластохинон))I Карнитнновые (карнитии)
Исходными веществами для образования коферментов первой группы являются витамины, поэтому недостаточное поступление их с пищей сразу сказывается на синтезе этих коферментов, а как • следствие нарушается и функция соответствующих сложных ферментов. Коферменты второй группы образуются из промежуточных продуктов обмена, поэтому недостатка в этих кофермелтах в физиологических условиях не бывает, и функция ферментов, с которыми они связаны, не нарушается.
| » < 4> |
| Коферменты оксидо-редуктаз (I) . Никотикамидяые (НАД, НАДФ) Флавиновые (ФМН, ФАД) Металлопорфириновые (reMi лорофиллы a, b Хинонкоферменгы (убихинон. пластсхи- он) Пептидные (глутатнон) Л] |
| . d). |
Существует и функциональная классификация коферментов, по которой коферменты относят, как и ферменты, к соответствующим шести классам (цифры в скобках указывают номер класса фермента):
Коферменты
Коферменты а
I Пиридоксиновые (ПАЛФ)
2. Пантотеновые (КоА, дефосфо-КоА)
3. Тиаминовые (ТДФ)
4. Кобамидные (дезоксиаденозилкобаламнн) Коферменты яэомераэ (5)
1. Пиридоксиновые (ПАЛФ)
2. Кобамидные (дезоксиаденозилкобаламнн)
3. Фосфаты моносахаридов (глюкозо-1, 6-ди- Коферменты трансфераз (2) фосфат. 2.3-дифосфоглииерат)
1 Пиридоксиновые (ПАЛФ, ПАМФ) 4 Пептидные (глутатнон)
2 Пантотеновые (КоА, дефосфо-КоА, 4-фос Коферменты лмгаа (6) фопантотенат) I Нуклеотидные (УДФ-глюкоза, ЦДф-холпн А. Нуклеотидные коферменты (УДФ-глюкоза, и т. д.)
ЦДФ-холнн и др) 2. Биотиновые (карбоксибиотии)
4 Птеридиновые, или фолиевые (ТГФК) 3. Фолневые (5, 10-метенил ТГФК)
5 Кобамидные (метилкобаламнн)
| II. Невитаминные кофермеиты 1 Нуклеотидные (УД Ф- глюкоз а, другие нук- леотидные производные углеводов, спиртов и т, д.) 2. фосфаты моносахаридов (глюкозо-1.6-ди- фосфат, 2.3 дифосфоглнаерат) 3. Металлопорфириновые (темы, хлоро- 4 Пептидные (глутатаон) |
Можно отметить две особенности коферментов. Первая — отсутствие коферментов третьего класса — гидролаз и полифункциональность ряда коферментов (пиридокснновых, кобамидных), т. е. способность одного и того же кофер мента катализировать разные реакции, в зависимости от того, в состав активного центра какого фермента он входит. Это служит наглядным примером значения апофермента в проявлении специфического участия кофермента в катализе.
Витаминные коферменты
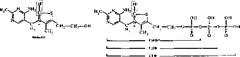
Тиаминовые коферменты. Источник их образования — тиамин (витамин В,), который по химическому строению относится к пиримидиновым производным тиазола. Наиболее активная коферментная форма его — тиаминдифосфат (ТДФ). Остальные производные тиамина—тиаминмонофосфат (ТМФ), тиамин-
|
трифосфат (ТТФ) считаются тоже коферментами, но значение их не выяснено ТДФ входит в состав ферментов, катализирующих окислительное декарбо- ксилирование а-кетокислот — пирувата и 2-оксоглутарата, а также является коферментом транскетолазы, осуществляющей превращение субстратов пенто- зофосфатного цикла. Причем «активным» участком в молекуле ТДФ, служащим местом присоединения субстрата, является атом углерода в кольце тиазола (заключен в рамку).
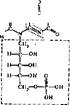
Флавинов'ые коферменты Источник их образования рибофлавин (витамин В2), который по химическому строению относится к производным нзоал- локсазина. Из рибофлавина синтезируются коферменты — флавин{4ононуклео- тид (ФМН) и флавинадениндинуклеотнд (ФАД):
" {н, " "
н—с—он
н—с—он I
н—с—он нхон
рибофлавин
|
Особенностью рибофлавина и его коферментов является способность к обратимым реакциям окисления — восстановления:
|
(здесь R — соответствующие радикалы, заключенные в предыдущих формулах в рамки).
Окисленные рибофлавин и оба кофермента имеют желтую окраску. При восстановлении они переходят в лейкоформу, и окраска раствора исчезает. Восстановленные коферменты ФМН • Н2 и ФАД ♦ Н2 образуются в резуль- тате присоединения атомов водорода к N-I и N-5 изоаллоксазннового кольца. Способность легко принимать и отдавать протоны и электроны определяет участие этих коферментов в окислительно-восстановительных реакциях.
|
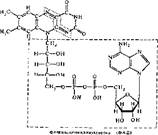
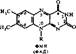
Пантотеновые коферменты. Пантотеновая кислота (витамин В3) служит исходным веществом для образования следующих коферментов: кофермента A (KoASH), дефосфокофермента А (дефосфо-КоА5Н), пантетеин-4-фос- фача (Пф), которые находятся з клетке в свободном виде или свячаны с ферментными белками. Коферменты участвуют в реакциях переноса ацильных групп. Отсюда и название — кофермент ацилирования (А). Кофермент А
H, 0—P—о—p—о—с н, —
сокращенно обозначается или KoASH, или просто КоА. Группа SH всех коферментов пантотеновой кислоты является рабочей, якорной частью молекулы. К ней присоединяются ацилы, и образуется метаболическая форма КоА — ацил~КоА (тиоэфирная связь — макроэргнческая, поэтому обозначается волнистой линией). Дефосфо-КоА и ПФ как коферменты используются меньше, чем KoASH. Считают, что дефосфо-КоА — кофермент, катализирующий расщепление цитрата, а Пф — кофермент ацилпереносящего белка син- тетазы жирных кислот.
| •со—n н—с llj—сн, - |
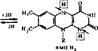
Никотинамидные коферменты. Источником образования ьикотинамидных коферментов является ниацин (витамин В5, РР, никотинамид). К никотина- мидным коферментам относятся никотинамидадениндинуклеотид (НАД) и
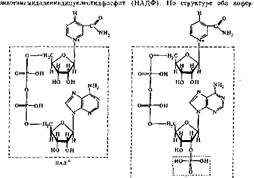
|
мента — динуклеотиды, в которых мононуклеотиды соединены фосфодиэфирной связью. В один из мононуклеотидов этих коферментов входит никотинамид; другой, представлен адениловой кислотой. В НАДФ имеется дополнительный остаток фосфорной кислоты, соединенный с гидроксилом рибозы.
Оба кофермента способны обратимо принимать электроны и протоны, поэтому они входят в состав дегидрогеназ. В реакциях, катализируемых ни- котинамидными ферментами, происходит отщепление двух атомов водорода от субстрата. Один атом водорода присоединяется к С-4 никотинамидного кольца; электрон второго атома водорода — к четвертичному азоту того же кольца, а оставшийся свободный протон переходит в среду. Окисленные формы коферментов сокращенно в реакциях обозначаются НАД+ и НАДФ+, а восстановленные — НАД ■ Н + Н" и НАДФ • Н + Н; (или упрощенно НАД • Н2 и НАДФ ■ На):
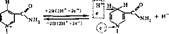 НАД* НАДН-^Н*
(надф+) (надф н+н4")
НАД* НАДН-^Н*
(надф+) (надф н+н4")
|
(здесь R — радикал, заключенный в приведенных выше соответствующих формулах в рамку).
Свободный витамин — никотинамид — не способен выполнять функции коферментов.
-Пиридоксиновые коферменты. Источником образования пиридоксиновых коферментов является пиридоксин (витамин В6). Это название объединяет три родственных витамина — пиридоксин, пиридоксаль, пиридоксамин
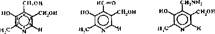
|
В клетках-организма из них образуются биологически активные коферменты пиридоксальфосфат (ПАЛФ) и пиридоксаминфосфат (ПАМФ):
н—с=о ch2nh2
палф памф
Первый из них является основным коферментом, входящим в состав многочисленных ферментов. Однако в некоторых реакциях ПАМФ выступает как самостоятельный кофермент, например в реакции образования 3, 6-дидезокси- гексоз, необходимых для синтеза гликопротеидов мембран бактерий.
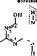
Фолиевые, или птериднновые, коферменты. Фолацин объединяет группу родственных витаминов, главный представитель которых — фолиевая кислота. В организме из нее образуется кофермент тетрагндрофолиевая кислота
•СО—NH—СН—СН, —С Н, —СООН
| ^С—С Н, —N Нт |
| лл^............ |
| ' i \=/ |
^оон
| 'I e^Hj |
участвующая в реакциях переноса следующих од ноу гле родных фрагментов, формила (—СН=0), формимина (—CH=NH), метенила (=СН), метила (—СН3) и метилена (=СН2) (в рамки заключены активные группы молекулы). ТГФК, связанная с одно углеродны ми радикалами, является метаболически активной формой.
| I i II |
Кобамидные коферменты. Источник образования кобамидных коферментов — витамин В, 2. Основная часть этого витамина — Со-комплекс азотистого макроцикла, называемого коррин. Коррин содержит четыре восстановленных пиррольных кольца, несущих различные заместители. Кобальт, находящийся в центре кольца коррина, может иметь различную степень окисления: от Со3+ до Со6+. Он связан ковалентной и координационными связями с атомами азота пиррольных колец коррина. В витамине В12 остальные связи заняты остатком 5, 6-диметилбензимидазолилриботида и группой CN. Поэтому витамин В|г называется цианокобаламином. Замена CN-группы на оксигруппу или ннтрогруппу приводит к образованию других витамеров В, 2 — соответственно оксокобаламина и нитриткобаламина. В организме он находится в виде ко- ферментных форм — метилкобаламина и 5-дезоксиаденозилкобаламина. Ниже приведено схематическое строение центральной части цианкобаламина (I) и его коферментных форм — метилкобаламина (II) и 5-дезоксиаденозилко
баламина (III):
—N-^! '" " N-
i ■ II
(здесь R — 5, 6-диметилбензимидазолилриботид, a R'— 5'-дезоксиаденозил). Эти коферменты участвуют в реакциях переноса групп, изомеризации и др.
Биотиновые коферменты. Биотин (витамин Н) образует активную кофер- ментную форму — карбоксибиотин:
----- _,
; НN__ _N Н'; HN _ _ _N_~ CO 0_ J
не—CH не—
H Г ^НГГНО СООН H.C СН(СН,)„СООН
'V •• " V
Ключевую роль в молекуле биотнна играют атомы азота, к которым присоединяется С02. Биотин участвует в переносе карбоксильных групп.
Липоевые коферменты. Исходным соединением для образования коферментов служит липоевая кислота (витамин N). Липоевые коферменты участ вуют в окислительно-восстановительных реакциях при превращении а-кето- кислот в пируватдегидрогеназном комплексе. Существуют окисленные и восстановленные формы липоевой кислоты, которая связана с ферментом (Е) амидной связью.

|
Хиноновые коферменты. Среди природных хиноидных соединений кофер- ментными свойствами обладает убихинон, или кофермент Q (KoQ), а также его аналог пластохинон, содержащийся в растительных организмах. Убихинон относят к липофильным витаминоподобным веществам. По химическому строению он представляет собой хинон с боковой изопреноидной цепью Число изопреноидных звеньев в.боковой цепи природных убихинонов разное, поэтому убихинопы обозначают символом Q„. В природе найдены убихиноны Q, — Q12. Наиболее часто встречаются коферменты Qs—Q10- Много убихинона содержится в мембранах митохондрий; имеется он также в мембранах эндо- плазматического ретикулума и ядер клеток. Убихинон способен к обратимым окислительно-восстановительным превращениям;
HjCO^JL
1 1 (тНз, :
НзСО^С н2-с н=с—С н^ н
При восстановлении он переходит в убихинол, который при окислении вновь превращается в убихинон. Благодаря окислительно-восстановительным свойствам убихинон участвует в переносе электронов и протонов в дыхательной цепи митохондрий, а его аналог пластохннон выполняет ту же роль в хлоро- пластах.
Для других природных хиноидных соединений — нафтохинонов (витамин К) и токоферолов (витамин Е), которые близки по строению и окислительно-восстановительным свойствам к убнхинону, коферментные функции пока не доказаны.
Карнитииовые коферменты. Битам иноподобное вещество карнитин (витамин Вт), являясь коферментом трансфераз, участвует в переносе ацильных групп (остатков уксусной кислоты и высших жирных кислот) через липидный слой митохондриальной, а возможно, и других мембран. Карнитин может находиться в развернутой и циклической формах:
Н3С-ч
| R-C-0-HC < • |
н, С'
Ацилы присоединяются к ОН-группе кар нити на с образованием соответствующего ацилкарнитина:
сн3
HjCv^fCH
-u-m. ^ „ _
О Н2 |
Н, С—N—СН3
< / ЧОН
Поскольку " циклическая форма более жирорастворима (вследствие экранирования зарядов метильными группами), то именно в такой, циклической, форме, как считают С. Е. Северин и др., карнитин способен диффундировать через липидный слой мембраны и переносить ацилы.
Невитаминные коферменты
Нуклеотидные коферменты. К нуклеотидным коферментам, не являющимся производными витаминов (этим они отличаются от рассмотренных нуклеотнд- ных коферментов — НАД, НАДФ, ФАД, КоА, в построении которых участ
вуют витамины), относятся нуклеозиддифосфаты и нуклеозидмонофосфаты, соединенные через концевой фосфат е различными субстратами.
Все нуклеотидные коферменты делятся на пять групп в зависимости от типа нуклеозида: уридиновые, цитиднновые, тимидиновые, аденозиновые и гу- анозиновые. Индивидуальные нуклеотидные коферменты внутри каждой группы отличаются друг от друга присоединенным к ним субстратом. Уже известно свыше 60 различных нуклеотидных коферментов, содержащих остатки Сахаров, спиртов, аминокислот, липидов, неорганических веществ. Наиболее представительна среди них группа нуклеозиддифосфатсахаров. Ниже приведено строение некоторых представителей нуклеотидных коферментов:
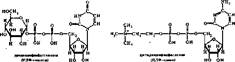
|
Большинство известных нуклеотидных коферментов представлено нуклео- зиддифосфатами; но встречаются н нуклеозидмонофосфаты, например ЦМф- сиаловая кислота. Реакции, в которых участвуют нуклеотидные невитаминные коферменты, можно разделить на два типа. К первому относятся реакции, связанные с превращением субстрата в молекуле кофермента. В этом случае кофермент уподобляется косубстрату. С субстратом в составе кофермента могут происходить различные, превращения: стерео изомеризация (например, УДФ-глюкоза превращается в УДФ-галактозу), окисление или восстановление (например, в печени происходит ферментативное окисление атома С6 глюкозы и превращение последней в УДФ-глюкуроновую кислоту) и т. д.
Реакции второго типа связаны с участием нуклеотидных коферментов как доноров субстратов в реакциях переноса групп. При этом происходит разрыв фосфоэфирных связей, соединяющих кофермент и субстрат. Этот тип реакций используется в синтезе различных веществ. Так, УДФ-глюкоза является донором глюкозы при биосинтезе гликогена, УДФ-глюкуроновая кислота — донор остатка глюкуроновой кислоты в реакциях конъюгации природных (например, билирубина) и чужеродных веществ, ЦДФ-холин — донор холина при биосинтезе холинфосфатидов и т. д.
Фосфаты углеводов как коферменты. Некоторые фосфаты углеводов выполняют функции коферментов. Так, глюкозо-1, 6-дифосфат выступает как кофермент фермента глюкозофосфатизомеразы, катализирующего обратимую изомеризацию глюкозо-6-фосфата и фруктозо-6-фосфата; 2, 3-дифосфоглице- рат — как кофермент фосфоглицератмутазы, участвующей в превращении 2-фосфоглицерата в 3-фосфоглицерат и обратно. Следует заметить, что 2, 3-дифосфоглицерат является также регулятором функций гемоглобина.
Металлпорфириновые коферменты. К ним относятся рассмотренные ранее гемы, которые участвуют как коферменты окислительно-восстановительных реакций, катализируемых оксидо-редуктазами (цитохромы, каталаза, перокси- даза, триптофаноксигеназа и др.)> и хлорофиллы, участвующие в окислительном разложении воды при фотосинтезе (см. гл. Образование энергия в фото- синтезирующнх организмах).
Пептидные коферменты. К ним относятся глутатион. По химическому строению это трипептид — глутаминнлцнстеинилглицин.
Функциональной группой его является SH-группа цистеина, которая способна к обратимым окислительно-восстановительным превращениям. Поэтому существует две формы глутатиона: восстановленная (сокращенно обозначается Г-SH) и окисленная, или дисульфндная (Г-S—ST):
НООС—СН—СН2—СН2— СО—NH—СН—СО—NH—СН*—СООН
I I
NH2 СН2
i„
НООС—СН—СН, —CHj— CO—NH—СН—со—NH—СН2—СООН
I I
NHj CHj
J
I
s
NH, U
I I
НООС—CH—CH5—CHj—CO—NH—CH—CO—NH—СНг—COOH
глутатион окныенньй ff-S-ST]
Глутатион — кофермент ряда оксидоредуктаз, например глутатионперо- ксидазы.
5. Ионы металлов как кофакторы ферментов
Кофакторами могут быть и ионы металлов. Мет алло ферменты — очень распространенная группа ферментов, составляющая '/< от всех ферментов. Роль металлов в этих ферментах различна. Металлоферменты делятся на две группы:
I. Ферменты, где ионы металлов выполняют роль активатора (эти ферменты могут катализировать и без металла).
II. Ферменты, где ионы металлов выполняют роль кофактора (без ионов металлов эти ферменты неактивны).
1) Диссоциирующие металлоферменты (ион металла легко диссоциирует от апофермента).
2) Недиссоциирующие металлоферменты (ион металла прочно связан с апоферментом):
а) теряющие активность при связывании металла реагентом;
б) не теряющие активности при связывании металла реагентом.
Ионы металлов как кофакторы входят в состав ферментов, относящихся к разным классам. Металлоферменты, катализирующие реакции окисления — восстановления, довольно многочисленны. Ион металла может находиться^
Таблица 21. Примеры метал лоферментов различных классов
|
в активном центре или входить в состав более крупной органической молекулы (например, гема), или может быть непосредственно связан с остатками аминокислот апофермента. Поскольку под действием оксидоредуктаз происходит перенос электронов и изменяется степень окисления субстратов, то в роли кофакторов выступают металлы с переменной валентностью: железо, медь, молибден, кобальт. v
Если металл непосредственно не участвует в катализе, а служит иным целям, например связывает субстрат, то в оксидоредуктазы входят металлы с постоянной степенью окисления.
Металлоферменты, катализирующие реакции гидролиза субстратов, содержат металлы с постоянной валентностью: цинк, кальций, магний. Редко в составе гидролаз находят металлы с переменной степенью окисления, например марганец (см. табл., 21).
Какова роль металлов в каталитическом действии фермента? Доказано несколько возможных вариантов участия ионов металла в работе фермента. Во-первых, металл, являясь своеобразной электрофильной группой активного центра, способен взаимодействовать с отрицательно заряженными группами субстрата. Такой металло-субстратный комплекс легче атакуется ферментом. Например, ионы Mg2+ (или Мп2+) образуют комплекс с АТФ или АДФ в реакциях, катализируемых креатинфосфокиназой и АТФазой. В результате этого активность фермента проявляется в полной мере, а в отсутствие металлов ферменты малоактивны или неактивны.
Во-вторых, металл с переменной валентностью сам может участвовать в транспорте электронов, т. е. выполнять функцию каталитического участка.
В-третьих, металл способствует формированию каталитически активной конформации третичной и четвертичной структуры апофермента. Стабилизация возможна путем образования солевых мостиков между ионом металла и карбоксильными группами кислых аминокислот при формировании третичной структуры белковой молекулы фермента или между субъединицами при образовании четвертичной структуры. Например, ионы кальция стабилизируют а-амилазу, а ионы цинка — алкогольдегидрогеназу. Лишенная цинка алко- гольдегидрогеназа диссоциирует на субъединицы и теряет активность.
В-четвертых, металлы иногда служат своеобразным мостиком между апоферментом и коферментом. Например, в алкогольдегндрогеназе ион цинка связывает НАД+.
Следует помнить, что так же как и коферменты витаминной природы, металлы поступают в организм с пищей. Поэтому нормальная функция большого семейства металлоферментов зависит от нормального поступления металлов, в большинстве своем относящихся к группе микроэлементов. Отсюда и высокая биологическая активность этих металлов: недостаточное поступление их с пищей может вызвать серьезные нарушения обмена веществ в организме.
6. Механизм действия ферментов
Сложная структурная и функциональная организация ферментов отчасти является ключом к пониманию характерных свойств ферментов — высокой специфичности и скорости катализа, не достижимой для неферментных катализаторов. Одной из первых гипотез, объясняющих действие ферментов, была адсорбционная гипотеза, предложенная в начале XX в. английским физиологом Бейлисом и немецким биохимиком Варбургом. При обосновании основных положений этой гипотезы исходили из механизма действия небиологнческнх катализаторов. Согласно адсорбционной гипотезе поверхность фермента, подобно губчатой платине, является местом адсорбции молекул реагентов. Тем самым облегчается их взаимодействие и реакция ускоряется. Однако эта гипотеза не объясняла специфичность действия ферментов и сейчас имеет лишь историческое значение.
Большую роль в развитии представлений о механизме действия ферментов сыграли классические работы Михаэлиса и Ментен, развивших положение о фермент-субстратных комплексах. Согласно представлениям Михаэлиса — Ментен весь процесс ферментативного катализа описывается простым уравнением (рнс. 21).
Процесс ферментативного катализа можно условно разделить на три стадии, каждая из которых имеет свои особенности.
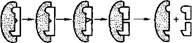 Е + S ■ а» ES ------- ES* -- ш- ES" ЕР -------------- Е + Р
Е + S ■ а» ES ------- ES* -- ш- ES" ЕР -------------- Е + Р
|
| •3= |
| / 2 3 Рис. 21. Схема стадий ферментативного катализа (Е — фермент, S — субстрат. Р — продукт) |
ч1. Диффузия субстрата к ферменту и стерическое связывание его с активным центром фермента (образование фермент-субстратного комплекса
2. Преобразование первичного фермент-субстратного комплекса в один или несколько активированных фермент-субстратных комплексов (обозначенных в уравнении ES* и ES**).
3. Отделение продуктов реакции от активного центра фермента н диффузия их в окружающую среду (комплекс ЕР диссоциирует на Е и Р).
Первая стадия, обычно непродолжительная по времени, зависит от концентрации субстрата в среде и скорости его диффузии к активному центру фермента. Образование комплекса ES происходит практически мгновенно. На этой стадии изменение энергии активации незначительно. Ориентация субстратов в активном центре фермента благоприятствует их сближению и прохождению реакции.
Вторая стадия наиболее медленная, и длительное 1ь ее зависит от энергии активации данной химической реакции. На этой стадии происходит расшатывание связей субстрата, их разрыв или образование новых связей в результате взаимодействия каталитических групп фермента. Именно'благодаря образований активированных переходных комплексов снижается энергия активации субстрата. Вторая стадия лимитирует скорость всего катализа.
Третья стадия непродолжительна, как и первая. Она определяется скоростью диффузии продуктов реакции в окружающую среду.
Молекулярные механизмы действия ферментов еще во многом неясны. Среди изученных механизмов действия ферментов можно отметить следующие:
1) эффект ориентации реагентов (сближения);
2) эффект деформации субстрата (напряжения, изгиба, натяжения);
3) кислотно-основной катализ;
4) ковалеитный катализ.
Эффект ориентации реагентов — очень характерное свойство ферментов, позволяющее ускорить превращение (повысить реакционную способность субстратов) в тысячи или десятки тысяч раз. Контактные участки актйаного центра фермента специфически связывают субстраты и обеспечивают их взаимную ориентацию и сближение так, чтобы это было выгодно для действия каталитических групп. Такая взаимная ориентация двух и более молекул, невозможная при беспорядочных соударениях в водной среде и на поверхности неорганического катализатора, способствует увеличению скорости реакции. Упорядоченное расположение субстратов приводит к снижению энтропии, а значит, способствует снижению энергии активации.
Эффект деформации субстрата (или так называемая теория «дыбы») хорошо объясняет действие гидролаз, лиаз и некоторых трансфераз. До присоединения к ферменту субстрат имеет «расслабленную» конфигурацию. После связывания с активным центром молекула субстрата как бы растягивается («напряженная», или «деформированная», конфигурация). Чем больше длина межатомной связи в субстрате, тем меньше энергия ее разрыва (т. е. снижается энергия активации). Места деформации (растяжения) легче атакуются, например молекулами воды.
Кислотно-основной катализ. Особенность активного центра фермента в отличие от других катализаторов состоит в том, что в нем имеются функциональные группы аминокислотных остатков, которые проявляют свойства как кислоты, так и основания. Поэтому фермент проявляет в ходе каталитического акта кислотно-основные свойства, т. е. играет роль и акцептора, и донора протонов, что невозможно для обычных катализаторов.
Таблица 22. Некоторые ферменты, обладающие способностью к ковалентному катализу
э продукта
. Химотряпсин, трипсин, тромбин, эстераза
2. Фосфоглюкомутаза, щелочная фосфатаза
-О—Р—О—CHj—СН—
| Ацилцнстеин R— С—S—С 11 О |
| HS—CHj—СН— |
| I |
3. Глицеральдегид-З-фосфатдегидрогеназа,
При закреплении субстрата в активном центре на его молекулу влияют электрофильные и нуклеофильные группы каталитического участка, что вызывает перераспределение электронной плотности на участках субстрата, атакуемого кислотно-основными группами. Это облегчает перестройку и разрыв связей в молекуле субстрата. Ярко выраженной способностью к кислотно- основному катализу обладают ферменты, в каталитическом центре которых имеется гистидин. Гистидин обладает отчетливыми кислотно-основными свойствами. При блокировании гистидина фермент инактивируется. Кислотно- основной катализ характерен для гидролаз, лиаз, изомераз. Он часто сочетается с ковалентным катализом.
Ковалентный катализ наблюдается у ферментов, которые образуют кова- лентные связи между каталитическими группами активного центра и субстратом. Ковадентные фермент-субстратные промежуточные продукты очень неустойчивы и легко распадаются, освобождая продукты реакции. В табл. 22 приведены некоторые ферменты, обладающие способностью к ковалентному катализу. Для большинства ферментов характерно сочетание описанных механизмов, что обеспечивает их высокую каталитическую активность.
7. Специфичность действия ферментов
Ферменты имеют разную специфичность и по отношению к субстратам. По степени специфичности ферменты делятся на следующие основные виды, упоминаемые в порядке снижения специфичности.
1. СтереохиМнческая субстратная специфичность — фермент катализирует превращение только одного из возможных стереоизомеров субстрата. Это крайний случай специфичности. Например, фумаратгидратаэа катализирует превращение только фумаровой кислоты (присоединение к ней молекулы воды), ко не ее стереоизомера — малеиниаой кислоты.
2, Абсолютная субстратная специфичность — фермент катализирует превращение только одного субстрата. Например, уреаза катализирует превращение только мочевины.
3. Абсолютная групповая субстратная специфичность — фермент катали-' зирует превращение сходной группы субстратов. Например, алкогольдегидро- геназа катализирует превращение не только этанола, но и других алифатических спиртов, хотя и с разной скоростью.
4. Относительная групповая субстратная специфичность — фермент специфически действует не на группу молекул субстрата, а на отдельные связи определенной группы субстратов. Например, пищеварительные ферменты — пепсин, трипсин — специфичны по отношению к пептидным связям, образованным определенными аминокислотами в разных белках.
5. Относительная субстратная специфичность — фермент катализирует превращение субстратов, принадлежащих к разным группам химических соединений. Например, фермент цнтохром участвует в гидроксилированин разных соединений (около 7000 наименований). Эти наименее специфичная ферментная система, участвующая в превращении природных веществ, лекарств и ядов.
Чем же объясняется специфичность действия ферментов? На этот счет, существует две точки зрения. Одна из них — гипотеза Э. Фишера, или, как ее называют, гипотеза «ключа и замка», или «шаблона», предусматривает, что в основе специфичности лежит строгое стерическое соответствие субстрата и активного центра фермента.
По Фишеру, фермент является жесткой структурой, активный центр которого представляет собой слепок субстрата. Если субстрат подходит к активно-
центру, как ключ к замку, то реакция произойдет. Если же субстрат («ключ») несколько изменен, то он не соответствует активному центру («замку»), и реакция становится невозможной. Гипотеза Фишера привлекает своей простотой в объяснении специфичности действия ферментов. Однако с позиций гипотезы «шаблона» трудно объяснимы, скажем, абсолютная и относительная групповая субстратная специфичность, поскольку слишком разнообразна конфигурация «ключей» (субстратов), которые подходят к одному и тому же «замку».
Объясняет эти внешние противоречия другая гипотеза, предложенная Кошлендом. Она получила название гипотезы «вынужденного соответствия». По мнению Кошленда, молекула фермента не жесткая, а гибкая, эластичная (что подтверждается современными методами исследования); конформация фермента и его активного центра изменяется при присоединении субстрата или других лигандов; и. наконец, активный центр — не жесткий слепок субстрата, а субстрат вынуждает его принять соответствующую форму в момент присоединения (отсюда и название гипотезы «вынужденного соответст вия»).
Иными словами, «замочная скважина», по Кошленду, сделана из податливого материала н поэтому принимает окончательную форму «ключа» при их контакте.
Гипотеза «вынужденного соответствия» получила экспериментальное подтверждение после того, как было зарегистрировано изменение расположения функциональных групп активного центра ряда ферментов после присоединения субстрата. Эта гипотеза позволяет также объяснить, почему происходит превращение близких аналогов субстратов. Если «ложный» субстрат (квазисубстрат) слабо отличается от природного и активный центр принимает конформацию. близкую к истинной, то расстановка каталитических групп в т

|
таком фермент-субстратном комплексе позволит осуществиться реакции (рис. 22). Этот «обман» фермент как бы не замечает. Однако ферментативная реакция пойдет не так быстро, как с истинным субстратом, поскольку нет идеального расположения каталитических групп в активном центре фермента.
Только в том случае, если конфигурация квазисубстрата не позволяет правильно разместиться каталитическим группам, то реакция не пойдет -(рис. 22, в). Очевидно, неодинаковая степень специфичности разных ферментов отражает как бы диапазон конформационных перестроек активного центра. Если он ограничен вплоть до единственно возможной конформации, фермент высоко специфичен. Если возможности перестройки велики, то фермент срабатывает и на квазисубстраты.
8. Кинетика ферментативных реакций
Кинетика действия ферментов — это раздел ферментологии, изучающий зависимость скорости реакции, катализируемой ферментами, от химической природы и условий взаимодействия субстрата с ферментом, а также от факторов среды. Иначе говоря, кинетика ферментов позволяет понять природу молекулярных механизмов действия факторов, влияющих на скорость ферментативного катализа.
Скорость ферментативной реакции определяется количеством вещестэа (или веществ), которое превращается в единицу времени. Скорость этих реакций зависит от влияния внешних условий (температуры, рН среды, влия ния природных и чужеродных соединений и т. д.).
Основы кинетики ферментативных реакций были заложены в работах Михаэлиса и Ментен. Скорость ферментативной реакции является мерой каталитической активности фермента и обозначается просто активность фермента. Измерить активность фермента можно только косвенно: по количеству превращаемого субстрата или нарастанию концентрации продукта в единицу времени.
Зависимость скорости ферментативной реакции от концентрации субстрата н фермента. Ферментативная реакция схематически описывается уравнением где k — константы скорости прямых (+) и обратных реакций (—).
Используя это уравнение, Бриггс и Холдейн вывели математическое выражение зависимости скорости реакции от концентрации субстрата:
v=iw[Si/(/C„+ [S]),
где v — наблюдаемая скорость реакции; — максимальная скорость реакции; Кт — константа Михаэлиса. Это уравнение называется уравнением
Михаэлиса — Ментен. При и = '/2итлх после соответствующих преобразований
та, т. е. Km= [S]. Следовательно, константа Михаэлиса имеет размерность концентрации.' Она равна концентрации субстрата, при которой скорость реакции равна половине макси- мальной, н выражается в молях на литр. Кт при k—x^k+t представляет собой или константу скорости ферментативной реакции Чем выше Кт, тем ниже скорость каталитического превращения субстрата данным ферментом. По значению Кт ферменты как бы можно условно разделить на «быстрые» (с низкой Кт) и «медленные» (с высокой Кт). Если какой-либо фермент катализирует двухсубстратную реакцию, то для каждого из субстратов есть своя Km, причем они могут значительно различаться. У ферментов с групповой субстратной специфичностью каждому субстрату соответствует своя Кт.
О сродстве субстрата к ферменту судят по субстратной константе, обозначаемой символом Ks- Она является константой диссоциации комплекса ES. Чем прочней связан субстрат, тем медленнее распадается ES на Е и S, а значит, такой субстрат имеет высокое сродство (специфичность связывания) к активному центру фермента и наоборот.
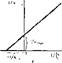
Графически зависимость скорости реакции от концентрации субстрата описывается гиперболой, называемой кривой Михаэлиса (рис. 23). Форма кривой показывает, что с увеличением концентрации субстрата все активные центры молекул фермента насыщаются. Это соответствует максимуму образования фермент-субстратных комплексов и максимальной скорости реакции vта» • Km легко находится на таком графике. Иногда график зависимости скорости реакции от концентрации субстрата строят методом двойных обратных величин (метод Лайнуивера — Берка) (рис, 23, 6). Значение константы Михаэлиса находят, как показано на графике.
От того, как изменяется скорость реакции при разных концентрациях субстрата, мож, но судить о порядке реакции, который необходимо знать для работы с ферментами и для правильного определения их активности в клинических лабораториях. Порядок реакции может варьировать от нулевого и выше. При нулевом порядке скорость реакции величина постоянная и не зависит от концентрации субстрата. При этом скорость реакции максимальна (Рпи*)- При первом порядке скорость реакции прямо пропорциональна концентрации одного из субстратов и т. д. Для того чтобы правильно определить активность фермента, нужно добиться нулевого порндка реакции, т. е. определять скорость ферментативно.й реакции при насыщающих концентрациях субстрата. В этом случае все изменения скорости реакции будут зависеть только от количества фермекга.
Чтобы оценить условия работы любого фермента в клетках организма, необходимо знать реально имеющиеся в них концентрации субстратов. В физиологических условиях ферменты почти никогда не работают в полную силу, ибо концентрации субстратов для них далеки от насыщающих. Пожалуй, только единственный субстрат, необходимый для гидролаз, — вода присутствует s клетках в насыщающих концентрациях, за исключением случаев, когда структурная локализация фермента ограничивает доступ воды к активному центру.
Зависимость скорости реакции от количества фермента носит линейный характер, что, как уже говорилось, отличает фермент от небиологических катализаторов. Из этого можно извлечь определенный практический вывод, что чем больше число молекул данного фермента в клетке организма по сравнению с остальными, тем выше в ней скорость химических превращений, катализируемых.этим ферментом. Если же какого-либо фермента недостаточно (нарушен синтез), то скорость реакции, катализируемая им, ограничивает ход связанных биохимических процессов.
Увеличение числа молекул фермента, достигаемое путем естественной стимуляции их образования или с помощью препаратов, позволяет или восстановить нарушенную скорость реакций, или приспособить необходимые биохимические реакции к новым условиям жизнедеятельности.
Зависимость скорости реакции от рН среды. Обычно кривая зависимости скорости ферментативной реакции от рЦ среды имеет колоколообразную форму (рис. 24), поскольку для каждого фермента существует свой оптимум рН, при котором скорость катализируемой им реакции максимальна. Отклонение рН в ту или другую сторону ведет к снижению скорости ферментативной реакции.
Оптимальные значения рН для некоторых ферментов
Фермент.. Пепсин Кислая Уреаза, Трипсин Аргиназа
фосфатаэа панкреатическая амнлаза
Оптимум рН 1, 5—2, 5 4, 5—5, 0 6, 4—7.2 7, 8 9.5—9.9
Из приведенных данных видно, что- оптимум рН у разных ферментов неодинаков. Однако большая часть ферментов клеток имеет оптимум рН, близкий к нейтральному, т. е, совпадающий с физиологическими значениями рН.
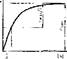 Рис. 23. Графики зависимости скорости ферментативной реакции от концентрации субстрата- а — Михаэлиса—-Ментен; б — Лайнуивера—Берна
Рис. 23. Графики зависимости скорости ферментативной реакции от концентрации субстрата- а — Михаэлиса—-Ментен; б — Лайнуивера—Берна
|
|
Зависимость скорости ферментативной реакции от рН главным образом свидетельствует о состоянии функциональных групп активного центра фермента. Изменение рН среды влияет на ионизацию кислых и основных групп аминокислотных остатков активного центра, которые участвуют или в связывании субстрата (в контактном участке), или в его превращении (в каталитическом участке). Поэтому специфическое влияние рН может быть вызвано
или изменением сродства субстрата к ферменту, или изменением каталитической активности фермента, или обеими причинами вместе.
Большинство субстратов имеют кислотные или основные группы, поэтому рН влияет на степень иониэа ции субстрата. Фермент предпочти тельно связывается или с ионизированной, или с неионизированной формой субстрата. Очевидно, при оптимальном рН и функциональные группы активного центра находятся в наиболее реакционноспособном состоянии, и субстрат находится в форме, предпочтительной для связывания этими группами фермента.
Зависимость ферментативной реакции от рН среды имеет практическое значение. Прежде всего определение активности ферментов должно проводиться при оптимальном для данного фермента рН. Для этого подбирается нужный буферный раствор с необходимым значением рН.
Диапазон колебаний рН в физиологических условиях незначителен, но изменения рН на ограниченном участке клетки могут быть. Они-то и влияют на деятельность ферментов. Например, при активной мышечной работе накапливается молочная кислота, кратковременно сдвигающая рН среды мышечных клеток в кислую сторону, что изменяет скорость ферментативных реакций. t
Знание оптимумов рН для индивидуальных ферментов важно для практической медицины. Например, пепсин для активного гидролиза белков в желудке требует сильнокислой среды, поэтому для восстановления нарушенной активности эндогенного пепсина необходимо принимать кислые вещества. Препарат пепсина принимают с соляной кислотои, создающей нужный рН.
|
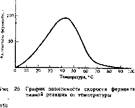
Зависимость скорости ферментативной реакции от температуры. С повышением температуры среды скорость ферментативной реакции увеличивается, достигая максимума при какой-то оптимальной температуре, а затем падает до нуля (рис. 25). Для химических реакций существует правило, что при повышении температуры на 10°С скорость реакции увеличивается в два-три раза. Для ферментативных реакций этот температурный коэффициент ниже: на каждые 10°С скорость реакции увеличивается в 2 раза и даже меньше. Наступающее вслед за этим снижение скорости реакции до нуля (нисходящая ветвь на рис. 25) свидетельствует о денатурации ферментного блока. Оптимальные значения температуры
для-большинства ферментов находятся в пределах 20—40°С. Термолабильность ферментов связана с их белковым строением. Некоторые ферменты денатурируют уже при температуре около 40°С, но основная часть их ни активируется при температурах выше 40—50°С. Отдельные ферменты инактивирует холод, т. е. при температурах, близких к 0°С, наступает денатурация.
Однако некоторые ферменты не подчиняются этим закономерностям. Так, фермент каталаза наиболее активен при температур ах, приближающихся к 0°С. Имеются и термостабильные ферменты. Например, аденилат- киназа выдерживает кратковременно температуру 100°С без инактивации. Микроорганизмы, обитающие в горячих источниках, содержат многие белки, в том числе и ферменты, отличающиеся высокой терм о стабильностью. Как уже упоминалось ранее, такие ферменты являются гликопротеидами, поскольку углеводный компонент придает белку термоустойчивость..
Влияние температуры на активность ферментов важно для понимания процессов жизнедеятельности. При снижении температуры некоторые животные впадают в состояние спячки или анабиоза. Скорость ферментативных реакций в этом состоянии замедляется, что обеспечивает малый расход накопленных организмом питательных веществ и снижение активности клеточных функций. Согревание тела ускоряет ход ферментативных реакций и возвращает организм животных к активной деятельности.
Искусственное охлаждение организма, так называемая гибернация, используется в клинике для проведения хирургических операций. Охлаждение тела также замедляет скорость ферментативных реакций, что позволяет снизить расход веществ и длительнее сохранять жизнеспособность клеток организма.
Повышение температуры тела (лихорадочное состояние), например при инфекциях, ускоряет биохимические реакции, катализируемые ферментами. Нетрудно подсчитать, что увеличение температуры тела на каждый градус повышает скорость реакции примерно на 20%. При высоких температурах около 39—40®С расточительное использование эндогенных субстратов в клетках больного организма обязательно требуется восполнять их поступлением с пищей. Кроме того, при температуре порядка 40°С часть весьма термолабильных ферментов может денатурироваться, что нарушает естественный ход биохимических процессов. Так что знание термозависимости ферментативных реакций позволяет использовать их в практической деятелкности врача.
Для определения активности ферментов в лабораторной практике всегда подбирают определенные стандартные или оптимальные температурные условия с учетом термолабильности конкретного фермента. Сравнивать изменения активности фермента, определяемого, скажем, для выявления нарушений в организме, можно только при одинаковых температурных режимах.
Термозависнмость ферментов используется в практике для разработки температурных режимов хранения продуктов питания. Сохранность их при низких температурах является результатом низкой активности собственных ферментов, которые «не съедают» свои субстраты (например, в овощах, фруктах и т. д.), или ферментов микроорганизмов, которые могут портить продукты.
9. Методы определения и единицы активности ферментов
Ферменты, содержащиеся в клетках, тканях и органах, предварительно экстрагируют, используя специальные методические приемы. При извлечении добавляют необходимые стабилизаторы ферментов, предохраняющие их от инактивации. Раствор фермента (экстракт из биологического материала) используют для определения ферментов. Сыворотка или плазма крови, другие биологические жидкости являются готовыми растворами ферментов, поэтому их сразу используют для определения. Если в задачу исследования входит получение очищенногч или кристаллического фермента, то определение активности ведут после каждой стадии очистки.
Качественные и количественные пробы на фермент проводятся косвенным путем по убыли субстрата или накоплению в среде продуктов реакции. Прямое измерение количества фермента в принципе возможно лишь для гомогенного, кристаллического фермента. Количество белка, измеряемого прямыми химическими методами, должно соответствовать количеству фермента. Практически же и в этом случае пользуются косвенным методом, так как количество белка в растворе гомогенного» фермента еще не критерий активности фермента (часть молекул может находиться в неактивном или денатурированном состоянии).
Скорость исчезновения субстрата или нарастания количества продуктов реакции в единицу времени служит мерой активности фермента.
Стандартные условия. Для правильного определения активности фермента необходимо проводить его в стандартных условиях, которые устанавливаются для каждого фермента из предварительных кинетических исследований, и точно измерять изменение содержания субстрата или продукта реакции за определенный отрезок времени.
Необходимо соблюдать оптимальное для определяемого фермента зна- 4eHHt рН (используют подходящий буфер). Концентрация субстрата должна быть больше насыщающей, при которой поддерживается максимальная скорость реакции (сверхнасыщающие концентрации субстрата специально устанавливаются' для ферментов, подверженных субстратному ингибирова- нию). Для сложных ферментов, требующих кофакторов (ионы металла, коферменты), концентрация кофакторов тоже должна превышать насыщающую. Стандартная температура принята 25°С (измерение при других температурах специально оговаривается в опыте). Эти стандартные условия обеспечивают нулевой порядок реакции, при котором изменение концентрации субстрата или продукта реакции зависит только от количества добавленного в среду фермента.
Для правильного измерения активности феБмента нужно определить начальную скорость реакции, т. е. в начале реакции, когда за равные промежутки времени пропорционально изменяется концентрация субстрата или продукта.
Методы определения содержания субстрата или продукта реакции. Определение проводится любым методом (колориметрическим, спектрофотомст- рическим, флуориметрическим, полярографическим и т. д.) после остановки реакции через определенный промежуток времени или регистрируется непрерывно в ходе реакции. Последний способ удобнее. Он возможен, если суб- 152 страт или продукт поглощают в определенной области спектра (регистрируют изменение поглощения их в ходе реакции на спектрофотометре) или флуоресцируют (изменение флуоресценции за определенное время непрерывно регистрируют на спектрофлуорнметрах) и т. д. Иначе говоря, выбор метода определения активности фермента ограничивается возможностями определения субстрата или Продуктов реакции.
Единицы активности ферментов. За международную единицу активности фермента принимается количество фермента, способное превратить один микромоль (мкмоль) субстрата за 1 мин в стандартных условиях. Международные единицы количества фермента обозначаются символом Е или U.
Удельная активность фермента равна массе фермента (r миллиграммах), способной превратить 1 мкмоль субстрата за 1 мин в стандартных условиях, выражается в мкмоль/(мин • мг белка"), Рекомендована также новая единица каталитической активности катал (символ — кат), которая представляет собой количество фермента, способное осуществить превращение 1 моля субстрата в 1 с " в стандартных условиях.
10. Регуляция активности ферментов
Ферменты, как уже говорилось, относятся к катализаторам с регулируемой активностью. Поэтому через ферменты можно контролировать скорость протекающих химических реакций в организме. Регуляция активности ферментов может осуществляться путем взаимодействия с ними различных биологических компонентов или чужеродных соединений (например, лекарств и ядов), которые принято называть модификаторами или регуляторами ферментов. Под действием модификаторов на фермент реакция может ускоряться (в этом случае их называют активаторами) или замедляться (в этом случае их называют ингибиторами).
Активация ферментов
Активация ферментов определяется по ускорению биохимических реакций, наступающему после действия модификатора. Одну группу активаторов составляют вещества, влияющие на область активного центра фермента. К ним относятся кофакторы ферментов и субстраты. Кофакторы (ионы металлов и коферменты) являются не только обязательными структурными элементами сложных ферментов, но и по существу их активаторами.
Ионы металлов бывают довольно специфичными активаторами. Часто для некоторых ферментов требуются ионы не одного, а нескольких металлов. Например, для Na^K^-АТФазы, осуществляющей транспорт одновалентных катионов через клеточную мембрану, необходимы в качестве активаторов ионы магния, натрия и калия.
Активация с помощью ионов металлов осуществляется по разным механизмам. В некоторых ферментах они входят в состав каталитического участка. В ряде случаев ноны металлов облегчают связывание субстрата с активным центром фермента, образуя как бы своеобразный мостик. Нередко металл соединяется не с ферментом, а с субстратом, образуя металлосубстрат- ный комплекс, который предпочтителен для действия фермента.
Специфичностью участия коферментов в связывании и катализе субстрата объясняется активация ими ферментативных реакций. Особенно заметно активирующее влияние кофакторов при действии на фермент, который не насыщен кофакторами.
Субстрат тоже в известных пределах концентраций является активатором. После достижения насыщающих концентраций субстрата активность фермента не возрастает. Субстрат повышает стабильность фермента и облегчает формирование нужной конформации активного центра фермента.,
Ионы металлов, коферменты и их предшественники и активные аналоги, субстраты можно использовать на практике как препараты, активирующие ферменты.
Активация некоторых ферментов может осуществляться путем модификации, не затрагивающей активный центр их молекул. Возможно несколько вариантов такой модификации: 1) активация неактивного предшественника — профермента, или зимогена; 2) активация путем присоединения какой- либо специфической модифицирующей группы к молекуле фермента; 3) активация путем диссоциации неактивного комплекса белок — активный фермент.
Ингнбнрование ферментов
Ингибиторы представляют большой интерес для понимания механизма ферментативного катализа. Применение различных веществ, связывающих функциональные группы контактного и каталитического участков активного центра фермента, может прояснить значение тех или иных групп, участвующих в катализе. Ингибиторы позволяют понять не только суть ферментативного катализа, но и являются своеобразным инструментом для исследования роли отдельных химических реакций, которые с помощью ингибитора данного фермента можно специфически выключать. Изучение ингибирования ферментативных реакций имеет прикладное значение для.изыскания и расшифровки механизма действия лекарственных средств, ядохимикатов и т. д.
Следует осторожно относиться к термину ингибитор, понимая под ним только то вещество, которое вызывает специфическое снижение активности фермента. Просто факт торможения реакции еще не говорит о том, что мы имеем дело с ингибитором. Любые денатурирующие агенты также вызывают угнетение ферментативной реакции. Поэтому в случае действия денатурирующих веществ правильнее говорить не об «ингибировании», а об «инактивации». Нередко вещество в небольших концентрациях является ингибитором, а в больших — инактиватором, так что деление это до некоторой степени условно..
Ингибиторы характеризуются прежде всего таким общим признаком, как прочность связывания с ферментом. По этому признаку ингибиторы делятся на две группы: обратимые и необратимые. Отнести ингибитор к одной из двух групп позволяет критерий восстановления активности фермента после диализа или сильного разведения раствора фермента с ингибитором. Необратимые ингибиторы прочно связываются с ферментом, и после этих процедур активность фермента не восстанавливается. Наоборот, комплекс фермент — обратимый ингибитор непрочен и быстро диссоциирует. Активность фермента при этом восстанавливается.
По механизму действия ингибчторы ферментов делятся на слеал-инике основные типы: 1) конкурентные: 2) неконкурентные; 3) бесконкурентныег 4) субстратные; 5) аллостерические
Конкурентным ингибированием называется торможение ферментативной реакции, вызванное связыванием с активным центром фермента ингибитора, сходного по структуре с субстратом и препятствующего образованию фермент-субстратного комплекса. При конкурентном торможении ингибитор и субстрат, будучи сходными по строению, конкурируют -за активный центр фермента. С активным центром связывается то соединение, молекул которого больше. С ферментом связан либо субстрат, либо ингибитор, поэтому для такого типа ингибирования справедливо уравнение
Е +1 — Е5
где I — ингибитор; EI — фермент-ингибиторныЙ комплекс. Но никогда при конкурентном ингибировании не образуется тройной комплекс ESI (фермент — субстрат — ингибитор), чем этот тип ингибирования отличается от других.
Ингибирование наступает вследствие того, что субстратоподобный ингибитор связывает часть молекул фермента, которые уже не способны образовать фермент-субстратный комплекс. Снять торможение можно избытком субстрата, вытесняющего ингибитор из активных центров ферментных молекул, тем самым возвращая им способность к катализу.
Вследствие сходства конкурентного ингибитора с субстратом такое ингибирование называют также изо стер ическим Конкурентными (изостери ческими.) ингибиторами могут быть метаболиты, накопление которых регулирует активность ферментов, и чужеродные вещества.
В качестве примера конкурентного ингибирования можно привести влияние различных веществ на активность сукцинатдегидрогеназы. Этот фермент входит в состав ферментной циклической системы — цикла Кребса. Его природным субстратом является сукцинат, а сходным с ним конкурентным ингибитором — оксалоацетат, промежуточный продукт того же цикла Кребса:
О
[]
-OOCt-CHJ— СН*—СОСГ -ООС—С—CHJ—СОО-
Аналогнчным конкурентным ингибитором сукцинатдегидрогеназы является малоновая кислота, часто, использующаяся в биохимических исследованиях. На рис. 26 схематически показан механизм конкуренции между сукцинатом и малонатом за фермент.
Наглядным примером конкурентного ингибирования может служить действие группы субстратов на ферменты, обладающие групповой субстратной специфичностью. Квазисубстраты являются конкурентными ингибиторами ферментов по отношению к истинным субстратам.
На принципе конкурентного ингибирования основано действие многих фармакологических препаратов, ядохимикатов, используемых для уничтожения сельскохозяйственных вредителей, и боевых отравляющих веществ.
Например, группа антихолинастеразных препаратов, к которым относятся производные четвертичных аммониевых оснований и фосфорорганнческие
Такие препараты, как прозе рин, физостигмин, севин, угне тают фермент обратимо, а фос
форорганические препараты типа армина, нибуфина, хлорофоса. 4арина, зома на действуют необратимо, фосфорилнруя каталитическую группу фермента В результате их действия накапливается ацетилхолин в тех синапсах, где он является медиатором нервного возбуждения, т. е. происходит отравление ор ганизма накопившимся ацетилхолин ом. Действие обратимых, ингибиторов постепенно проходит, так как чем больше накапливается ацетиЛхолина, тем быстрее он вытесняет ингибитор из активного центра холинэстеразы. Токсичность необратимых ингибиторов несравненно выше, поэтому их применяют для борьбы с вредителями сельского хозяйства, бытовыми насекомыми и грызунами (например, хлорофос) и как боевые отравляющие вещества (например, зарин, зоман н др.).
Избирательно выключая тот или иной фермент, можно проводить своеобразный анализ участия конкретного фермента в обмене веществ. Явление конкурентного ингибирования открывает возможности для поиска антиметаболитов, которые, обладая сходной конфигурацией с истинным субстратом, могут попадать в категорию конкурентных ингибиторов. Антиметаболи- ты перспективны как специфические фармакопрепараты.
Однако нельзя забывать, что конкурентные отношения возможны не только между субстратом и ингибитором, но и между ингибитором и кофер- ментом.
Антикоферменты (аналоги коферментов, не способные выполнять их функцию) тоже действуют как конкурентные ингибиторы, выводя «из строя» те молекулы фермента, с которыми они соединяются. Антикоферменты (или их предшественники антивитамины) широко используются и в биохимических исследованиях, и в медицинской практике как эффективные лекарства.
|
| Ацетнлхолин +■ НгО |
| Рис 26. Схема конкурентного ингнбирояания сукин н атд еги дрог е н азы м ал он атом |
| соединения, являются конкурентными ингибиторами фермента хо- линэстеразы по- отношению к его субстрату ацетилхолину. Холин- эстераза катализирует гидролиз ацетилхолина — медиатора хо- линэргических систем " (нервио- мышечных синапсов, парасимпатической системы и т. д.): |
| -Холин +■ Ачетат |
| Антихолинэстеразные вещества конкурируют с ацетил холи ном за активный центр фермента, связываются с ним и выключают каталитическую активность фермента. |
Неконкурентным иигибированием ферментов называется торможение, связанное с влиянием ингибитора на каталитическое превращение, но не на связывание субстрата с ферментом. Неконкурентный ингибитор или связывается непосредственно с каталитическими группами активного центра фермента, " или, связываясь с ферментом вне активного центра, изменяет конфор-
мацию активного центра таким образом, что затрагивает структуру каталитического участка, мешая взаимодействию с ним субстрата. Поскольку неконкурентный ингибитор не влияет на связывание субстрата, то в отличие от конкурентного ингибирования наблюдается образование тройного комплекса ESI по уравнению
Е + S + I — ESI
Однако превращения этого комплекса в продукты не происходит.
Неконкурентными ингибиторами являются, например, цианиды, которые прочно соединяются с трехвалентным железом, входящим в каталитический участок гемннового фермента — цитохромоксидазы. Блокада этого фермента выключает дыхательную цепь, и клетка, погибает. К неконкурентным ингибиторам ферментов относятся ионы тяжелых Металлов и их органические соединения. Поэтому ионы тяжелых ме
|
|