Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Основные сведения о методе капиллярного электрофореза
|
|
Капиллярный электрофорез
Цель работы
Изучение возможностей метода капиллярного электрофореза при определении неорганических анионов в водопроводной воде.
Основные сведения о методе капиллярного электрофореза
Метод капиллярного электрофореза основан на разделении заряженных компонентов сложной смеси в кварцевом капилляре под действием приложенного электрического поля. Микрообъем анализируемого раствора (~2 нл) вводят в кварцевый капилляр, предварительно заполненный подходящим буфером — электролитом. После подачи высокого напряжения (до 30 кВ) к концам капилляра компоненты смеси начинают двигаться с разной скоростью, зависящей, в первую очередь, от заряда и массы (точнее, величины ионного радиуса) и, соответственно, в разное время достигают зоны детектирования. Полученная последовательность пиков называется электрофореграммой; качественной характеристикой вещества является время миграции, а количественной — высота или площадь пика, пропорциональная концентрации вещества.
Для того чтобы получить более подробное представление о методе, необходимо рассмотреть ряд процессов, происходящих в капилляре, заполненном электролитом и помещенном в продольное электрическое поле.
Находящиеся на поверхности плавленного кварца силоксановые группы при контакте с водой или водными растворами гидролизуются с образованием удвоенного количества силанольных групп, которые затем гидратируются.
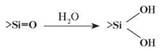
Скорость и степень гидролиза зависят от температуры и pH водных растворов и, в меньшей степени, от концентрации солевого фона раствора. В водном растворе силанольные группы способны к кислотной диссоциации. Константа первой ступени имеет величину Ка1 = 2, 5x10 3. Это означает, что при pH водного раствора больше 2, 5 поверхность кварца приобретает некоторый отрицательный заряд, который возрастает при увеличении pH раствора. Наоборот, при pH ~2 и меньше диссоциация сила- нольных групп практически полностью подавлена, и поверхность кварца становится нейтральной.
Диссоциация силанольных групп вызывает на границе раздела кварц—водный раствор электролита образование двойного электрического слоя (ДЭС), рис. 1. Первую его обкладку составляют неподвижные отрицательно заряженные силанольные группы. Вторую обкладку двойного слоя составляют положительно заряженные катионы, существующие в растворе. Диэлектриком, разделяющим обкладки этого конденсатора, являются молекулы воды, гидратирующие как силанольные группы, так и катионы.
Положительная часть ДЭС, в свою очередь, делится на две части: первую (или неподвижную), непосредственно примыкающую к поверхности кварца, и вторую (или диффузную), располагающуюся на некотором удалении от поверхности. В неподвижной части количество положительных зарядов меньше, чем отрицательных зарядов на поверхности кварца из-за увеличения размеров катионов вследствие гидратации. В результате в диффузной части ДЭС образуется некоторая избыточная концентрация катионов. Между этими двумя слоями проходит т. н. граница скольжения — при наложении вдоль капилляра электрического поля неподвижная часть остается на месте, в то время как диффузная часть начинает мигрировать к катоду, увлекая за собой в силу межмолекулярного сцепления всю массу жидкости в капилляре. Возникает электроосмотический поток (ЭОП), который осуществляет пассивный перенос раствора внутри капилляра. Скорость ЭОП в сильной степени зависит от pH раствора: в сильнокислых растворах ЭОП отсутствует, в слабокислых — его скорость незначительна, а при переходе в нейтральную и щелочную область pH скорость ЭОП возрастает до максимально возможной. С другой стороны, эта величина зависит от концентрации электролита в ведущем буфере: чем она больше, тем выше становится доля катионов в неподвижной части ДЭС, а толщина диффузной части уменьшается и, соответственно, уменьшается скорость электроосмотического потока.
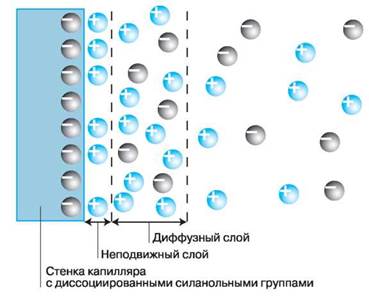
Рис. 1. Строение двойного электрического слоя.
В приборах для капиллярного электрофореза капилляр, заполненный раствором электролита, своими концами опущен в два содержащих тот же электролит сосуда, в которые введены электроды. Электролит должен обладать буферными свойствами, чтобы, с одной стороны, воспрепятствовать изменению состава раствора в приэлектродных пространствах, а с другой — стабилизировать состояние компонентов пробы в процессе анализа. При подаче на электроды высокого напряжения в капилляре быстро устанавливается стационарное состояние: через капилляр протекает постоянный электроосмотический поток, на который накладывается взаимно противоположная электромиграция катионов и анионов.
Если в капилляр со стороны анода ввести небольшой объем раствора пробы, то ЭОП будет переносить эту зону к катоду (в область детектирования), и зона некоторое время сможет находиться в капилляре под воздействием электрического поля высокого напряжения. В течение этого времени заряженные компоненты пробы будут перемещаться в соответствии с их электрофоретическими подвижностями.
Катионные компоненты пробы, двигаясь к катоду, будут обгонять электроосмотический поток (рис. 2). Скорость их движения складывается из скорости ЭОП и скорости электромиграции, поэтому на выходе капилляра катионы появляются первыми и тем раньше, чем больше их электрофоретическая подвижность.
Нейтральные компоненты пробы способны перемещаться только под действием электроосмотического потока, тогда как анионные будут перемещаться к аноду со скоростями меньшими, чем скорость ЭОП. Медленно мигрирующие анионы появятся на выходе после ЭОП, а те, чья скорость электромиграции по абсолютной величине превышает скорость ЭОП, будут выходить из капилляра в прианодное пространство.
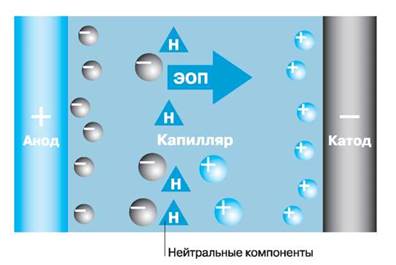
Рис. 2. Электрофоретическая миграция ионов в присутствии электроосмотического потока.
Если время нахождения пробы в капилляре (которое можно регулировать изменением напряжения, величины pH и концентрации ведущего электролита) достаточно, чтобы проявились различия в подвижности ионов, то на выходе капилляра вблизи катода можно наблюдать зоны раствора, в которых находятся индивидуальные компоненты пробы.
Ведущий электролит (его называют также рабочим буферным раствором) должен иметь такую концентрацию, при которой электрическое сопротивление раствора в капилляре будет достаточно велико. Это требование связано с тем, что при прохождении электрического тока в проводнике выделяется тепло. Если ток достаточно велик, то жидкость в капилляре может даже закипеть.
Основные варианты капиллярного электрофореза
Наиболее распространенными вариантами метода капиллярного электрофореза являются капиллярный зонный электрофорез и мицеллярная электрокинетическая хроматография.
Самым простым вариантом КЭ является капиллярный зонный электрофорез (КЗЭ). Компоненты сложной смеси движутся в среде электролита с разными скоростями, образуя дискретные зоны. Отличительная особенность КЗЭ состоит в том, что он пригоден для разделения только ионогенных компонентов пробы, тогда как нейтральные соединения, не обладающие собственной электрофоретической подвижностью, движутся со скоростью ЭОП и выходят в зоне нейтральных компонентов, зоне маркера ЭОП.
В приборах капиллярного электрофореза, в которых используется кварцевый капилляр, полярность входного конца чаще всего положительная (анод), и ЭОП переносит зону пробы к катоду. Вблизи катодного выхода установлен детектор. При этих условиях катионные компоненты пробы, тоже мигрируя к катоду, обгоняют ЭОП и первыми достигают детектора в виде отдельных зон, которые на электрофореграмме регистрируются индивидуальными пиками. Через некоторое время детектора достигает и зона исходного раствора, в которой остались нейтральные компоненты пробы. В зависимости от того, поглощают они или нет, на электрофореграмме регистрируется прямой (в некоторых случаях обратный) пик, который часто называют системным. Иногда для идентификации системного пика в пробу добавляют специальные вещества — маркеры ЭОП, например, бензиловый спирт. Что касается анионных компонентов пробы, то их поведение зависит от соотношения скоростей ЭОП и электромиграции анионов. Если скорость миграции аниона превышает скорость ЭОП, то такой анион рано или поздно выйдет из капилляра в прианодное пространство (это нежелательно, т. к. некоторые анионы, например хлорид, попадая в рабочий буферный раствор, будут, разряжаясь на аноде, вызывать коррозию платинового электрода). Если же скорость электромиграции аниона меньше скорости ЭОП, то такой анион может быть зарегистрирован на той же электрофореграмме после выхода системного пика. В этом варианте КЗЭ с положительной полярностью могут определяться катионные компоненты проб и большинство органических анионов.
Чтобы методом КЗЭ можно было определять анионные компоненты проб (в основном, неорганического происхождения) необходимо изменить полярность прикладываемого напряжения. Однако в этом случае изменится не только направление миграции анионов, но также направление ЭОП. Для преодоления этого противоречия необходимо модифицировать поверхность кварцевого капилляра так, чтобы знаки зарядов двойного электрического слоя поменялись на обратные. Это достигается введением в рабочий буферный раствор катионного поверхностно-активного вещества, например, бромида цетилтриметиламмония (ЦТАБ). Катион ЦТА+ активно сорбируется на кварцевой поверхности, занимая при достаточной его концентрации все вакансии в ближайшем к поверхности слое. Поверхность как бы «ощетинивается» длинными цетильными (С16Н33—) цепочками. Ставшая гидрофобной поверхность при дальнейшей промывке рабочим буферным раствором сорбирует еще один слой поверхностно-активного катиона, ориентированного аммонийным концом наружу (сорбция «щетка в щетку»). В результате первый слой двойного электрического слоя становится положительным, а второй, в том числе и диффузная его часть, — отрицательным, и ЭОП снова движется от входного конца к детектору, несколько отставая от мигрирующих быстрее анионов.
Основным достоинством КЗЭ является высокая эффективность (сотни тысяч теоретических тарелок), при этом селективность, определяемая механизмом разделения внутри одной фазы, в КЗЭ недостаточна. Повышение селективности может быть достигнуто за счет изменения pH ведущего электролита, введения в состав буфера различных добавок: поверхностно-активных веществ, макроциклов, органических растворителей и т. д.
Мицеллярная электрокинетическая хроматография объединяет электрофорез и хроматографию. МЭКХ получила наиболее широкое распространение среди других вариантов капиллярного электрофореза, в первую очередь, за счет способности разделять как ионогенные, так и незаряженные компоненты пробы. Разделение нейтральных соединений стало возможным благодаря введению в состав ведущего электролита поверхностно-активных веществ (ПАВ) — мицеллообразователей. Чаще всего используют анионные ПАВ (например, додецилсульфат натрия — ДДСН, англ. SDS) в концентрациях, превышающих критическую концентрацию мицеллообразования (ККМ), которая, например, для ДДСН в водном растворе составляет 8 мМ. В этом случае в растворе электролита находятся преимущественно мицеллы и небольшая доля мономерной формы ПАВ. Мономеры состоят из гидрофобного «хвоста» и гидрофильной (в случае анионного по- верхностно-активного вещества отрицательно заряженной) «головы». При формировании прямых мицелл мономерные фрагменты агрегируются неполярными концами внутрь, а внешняя сферическая поверхность мицеллы становится отрицательно заряженной. Каждая мицелла окружена собственным двойным электрическим слоем, внешнюю диффузную часть которого формируют катионы, присутствующие в растворе ведущего электролита. Число мономеров, образующих мицеллу, может колебаться от 60 до 100 молекул, однако общий заряд мицеллы существенно меньше из-за наличия в неподвижной части второго слоя ДЭС гидратированных катионов. Ни мицеллярная, ни мономерная форма АПАВ не взаимодействуют со стенкой кварцевого капилляра, но при подаче на капилляр высокого напряжения обе формы мигрируют к аноду, в то время как ЭОП направлен к катоду. Если в капилляр на анодной стороне ввести пробу, содержащую нейтральные и заряженные компоненты, то ЭОП будет переносить их к катоду, а навстречу будет двигаться поток отрицательно заряженных мицелл АПАВ. Нейтральные компоненты пробы могут распределяться между фазой раствора и мицеллярной фазой, причем константа этого распределения специфична для каждого сорта молекул пробы. В результате на выходе капилляра регистрируется электрофореграмма нейтральных компонентов, а также медленно мигрирующих анионов пробы.
Общее устройство систем КЭ
Минимальный состав системы, реализующей метод капиллярного электрофореза, должен включать следующие узлы: кварцевый капилляр, источник высокого напряжения, устройство ввода пробы, детектор и систему сбора, обработки и вывода информации (рис. 3).
Дополнительными устройствами в системах капиллярного электрофореза являются, например, автосемплер и блок жидкостного охлаждения капилляра, которые позволяют:
► автоматизировать подачу образцов,
► осуществить эффективный отвод тепла от капилляра.
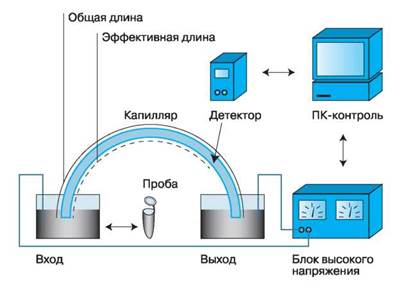
Рис. 3. Устройство системы капиллярного электрофореза.
Капилляры
В системах капиллярного электрофореза используют, как правило, капилляры из высокочистого плавленого кварца, прозрачного в УФ-области спектра, с внешним полимерным, чаще полиимидным, защитным покрытием. В случае детектирования внутри капилляра (on-line) полиимидное покрытие в зоне детектирования снимают, оставляя для прохождения света зону чистого кварца. Внутренний диаметр капилляров может варьироваться от 20 до 100 мкм, но чаще всего используют 50 и 75 мкм. Внешний диаметр составляет 365 мкм, длина капилляров 20—100 см.
Доминирующее число разделений в КЭ ведут на непокрытых изнутри капиллярах, так называемых немодифицированных. Их подготовка к анализу начинается, как правило, с промывки раствором щелочи для обеспечения диссоциации силанольных групп кварца и возникновения ЭОП.
Анализ методом КЭ можно проводить только тогда, когда капилляр находится в кондиционном состоянии. С точки зрения анализа кондиционное состояние капилляра следует понимать так, что выполняемые последовательно анализы должны быть воспроизводимы как по временам миграции пиков, так и по площадям пиков. При подготовке к работе капилляр обычно промывают раствором кислоты, водой и раствором щелочи. Цель первой операции заключается в удалении с поверхности примесей, в частности, многовалентных катионов, и первичном гидролизе силокса- новых групп. Промывка водой способствует удалению кислоты и дальнейшему гидролизу поверхности. Наконец, щелочная промывка предназначена для удаления примесей, не реагирующих с кислотой, и максимальной диссоциации образовавшихся силанольных групп. Финишная промывка водой имеет целью удалить из капилляра щелочь.
Источники высокого напряжения
В первую очередь, источники напряжения должны обеспечивать регулируемую подачу напряжения в диапазоне от —25 до +25 кВ и при заданной величине напряжения поддерживать постоянство этого значения. Максимально допустимый ток в капилляре при этом не должен превышать 200 мкА.
Как правило, переключение полярности происходит в ручном режиме, что сопровождается сменой высоковольтных блоков.
Ввод пробы
Типичный объем вводимой пробы в капиллярном электрофорезе составляет 1—20 нл. Общепринято заполнять пробой не более 2 % объема капилляра с тем, чтобы изначально, до анализа, не создавать широкую зону компонентов и обеспечить достаточное время нахождения зоны пробы в капилляре для установления значимых различий в электрофоретических подвижностях.
Непосредственно перед вводом пробы капилляр промывают рабочим буферным раствором, удаляя остатки пробы от предыдущего ввода.
Различают три способа ввода пробы:
► гидродинамический,
► электрокинетический,
► гидростатический.
Первые два способа реализованы во всех коммерческих системах капиллярного электрофореза, гидростатический, напротив, не нашел широкого применения.
Ввод пробы давлением (гидродинамический, пневматический) обеспечивается созданием разницы давлений между сосудом для пробы и выходным концом капилляра, при этом давление либо повышается в сосуде для пробы, либо снижается на конце капилляра.
Электрокинетический ввод пробы. При этом способе ввод пробы осуществляется путем подачи высокого напряжения на электроды, когда на входе установлена пробирка с раствором пробы, а на выходе — с рабочим буфером. За счет возникающего при этом ЭОП компоненты пробы перемещаются в капилляр. Количество введенной пробы при этом способе зависит от величины приложенного напряжения, времени, в течение которого приложено напряжение, и подвижности компонентов пробы.
Гидростатический ввод пробы. В этом способе для ввода пробы используют разницу в высоте между буферным сосудом и сосудом для проб.
Детекторы
Детектирование в системах капиллярного электрофореза может осуществляться различными способами:
► непосредственно в капилляре в части, близкой к выходному концу, в режиме реального времени (on-capillary). В зоне детектирования с внешней стенки капилляра снимают защитное полиимидное покрытие. Этот способ характерен для большинства коммерческих систем капиллярного электрофореза;
► непосредственно на выходном конце капилляра (end-capillary)',
► вне системы КЭ (off-capfflary, при этом, как правило, детектор представляет собой отдельный самостоятельный прибор (например, масс-спектрометр) и соединен с системой капиллярного электрофореза специальным интерфейсом).
Основными принципами детектирования в КЭ являются:
► фотометрическое в УФ-видимой области спектра (прямое и косвенное),
► флуориметрическое (прямое и косвенное),
► масс-спектрометрическое,
► кондуктометрическое,
► амперометрическое (прямое и косвенное),
► радиометрическое,
► рефрактометрическое.
Наиболее распространенным вариантом детектирования продолжает оставаться фотометрическое, основанное на поглощении веществом УФ или видимого света. Фотометрические детекторы в КЭ, подразделяют на:
► Детекторы с фиксированной длиной волны: источники света с линейчатым спектром (ртутная лампа (254 нм), кадмиевая лампа (229 нм) и цинковая лампа (214 нм). В приборах «Капель-104» фотометрический детектор работает на длине волны 254 нм (строго 253, 7 нм), поэтому отклик детектора будет наблюдаться только в том случае, если определяемый компонент имеет заметное поглощение на указанной длине волны
► Детекторы с изменяемой длиной волны: источниками света служат дейтериевые и вольфрамовые лампы (рабочий диапазон длин волн 190—350 нм и 340—850 нм, соответственно). Необходимая спектральная селекция достигается применением монохроматоров или узкополосных светофильтров.
► Детекторы на диодной матрице (ДМД). В таких детекторах световой поток, прошедший через капилляр, разлагается в спектр с помощью высококачественного светосильного монохроматора, а матрица фотодиодов постоянно регистрирует сигналы в ультрафиолетовой и видимой частях спектра (УФ-В-детекторы), обеспечивая запись в режиме сканирования. Данные, полученные одновременно на различных длинах волн (до 5), обрабатываются с помощью компьютеров, выделяющих сигнал на оптимальной длине волны и вычитающих фон. Применение детекторов на диодной матрице обеспечивает получение аналитических данных с гораздо большей степенью достоверности.
Для соединений, анализируемых с помощью КЭ и не поглощающих в УФ-диапазоне, существует возможность регистрации методом косвенного УФ-детектирования. В этом случае в состав ведущего электролита вводят небольшое количество хромофора — вещества, поглощающего на требуемой длине волны. Так, в случае определения анионов поглощающий ион тоже должен быть анионом, например, хромат-ион, фталат-ион, а при определении катионов чаще всего используют катионы ароматических аминов или гетероциклов, в частности, ион протонированного бензимидазола. Так как ионная сила ведущего электролита в процессе разделения остается постоянной, в зоне, где находится непоглощающий ион, уменьшается концентрация поглощающего иона. Обмен происходит строго эквивалентно, на электрофореграмме наблюдаются обратные (отрицательные) пики, площади которых пропорциональны концентрациям определяемых ионов. Косвенное УФ-детектирование является универсальным вариантом детектирования, т. к. позволяет регистрировать все присутствующие в анализируемом растворе компоненты.
Капиллярный электрофорез относится к группе комбинированных методов анализа, в которых объединены два основных процесса: предварительное разделение компонентов сложной смеси и их определение/детектирование. Важными характеристиками разделения являются разрешение, эффективность и селективность. Для конечного определения наиболее актуален параметр чувствительности, в первую очередь зависящий от типа используемого детектора.
Метод капиллярного электрофореза характеризуется высокой эффективностью.
Несмотря на высокую эффективность, достигаемую в капиллярном электрофорезе, селективность разделения, особенно в зонном варианте может быть недостаточна, в первую очередь, из-за осуществления процесса разделения внутри одной фазы. Задача повышения селективности разделения в том или ином варианте КЭ требует знания факторов, ее определяющих, и может быть решена за счет изменения pH ведущего электролита, введения в состав буфера различных добавок, например, ПАВ, макроциклов, органических. Следует иметь в виду, что все эти факторы будут сказываться также на скорости ЭОП, однако, сам по себе электроосмотический поток не ответственен за изменение селективности разделения и определяет лишь изменение времени миграции (на равную величину для всех компонентов пробы).
Выбор ведущего электролита является чрезвычайно важной задачей для успешного разделения в любом варианте КЭ. Величина pH ведущего электролита определяет как скорость течения жидкости в капилляре (величину ЭОП), так и форму нахождения компонента в растворе (заряд). Чувствительность ЭОП к изменению pH раствора заставляет использовать ведущие электролиты с высокой буферной емкостью, при этом диапазон pH, как правило, имеет значения рКа±1. Благодаря высокой стабильности кварцевого капилляра при электрофоретическом разделении можно использовать буферные системы с pH от 2 до 12.
Идеальный буфер для капиллярного электрофореза должен обладать следующими свойствами:
► достаточная буферная емкость в выбранном диапазоне pH,
► малое поглощение на длине волны детектирования,
► низкая подвижность ведущего иона.
Список так называемых «подходящих» буферов возглавляют боратный буфер и TRIS, так как они могут использоваться в широком диапазоне концентраций без существенного увеличения тока, что позволяет, в свою очередь, применять максимально высокие напряжения в ходе анализа.
Среди используемых в капиллярном электрофорезе добавок наиболее популярны поверхностно-активные вещества. Их введение в состав буферных растворов позволяет в разной степени влиять на селективность, причем определяющими факторами являются тип ПАВ и его концентрация. В КЭ могут быть использованы как ионогенные (катионные (КПАВ) и анионные (АПАВ), а также цвиттер-ионные), так и нейтральные поверхностно-активные вещества.
При концентрации ниже ККМ мономерные формы ионогенных ПАВ могут выступать как ион-парные добавки (различные АПАВ, КПАВ), а также влиять на растворимость гидрофобных компонентов смеси и модифицировать стенки капилляра (например, ЦТАБ). Возможные при этом механизмы взаимодействий поверхностноактивного вещества и пробы — ионные и/или гидрофобные. Добавки ПАВ в ведущий электролит влияют не только на поведение зоны пробы в капилляре, но и на стенки самого капилляра, модифицируя ЭОП (уменьшая, увеличивая или обращая).
Органические растворители (метанол, ацетонитрил, изопропанол и др.), которые вводят в буферный раствор в концентрации от нескольких долей процента до 30 % (об.) могут, с одной стороны, повышать растворимость анализируемых соединений, делая капиллярный электрофорез пригодным для анализа веществ с ограниченной растворимостью в водных средах. С другой стороны, органические добавки могут уменьшать гидрофобные взаимодействия между анализируемым компонентом и мицеллой в МЭКХ, а также влиять на подвижность ЭОП и собственную электрофоретическую подвижность аналита. Макроциклические реагенты как компоненты ведущих электролитов широко распространены в КЭ.
Обработка результатов в капиллярном электрофорезе. Качественный и количественный анализ
Целью любого анализа является получение ответов на вопросы: какие компоненты присутствуют в анализируемом образце и какова величина их концентраций? Первый из вопросов есть задача качественного анализа, второй — количественного. Для решения обеих задач в КЭ перед анализом пробы обязательно проводят процедуру градуировки системы путем измерения одной или нескольких смесей с известным качественным и количественным составом. Результатом градуировки являются формирование таблицы компонентов (содержит времена миграции и имена определяемых компонентов) и построение градуировочной зависимости (показывает зависимость сигнала детектора от концентрации/содержания вещества).
В капиллярном электрофорезе используют те же принципы интегрирования пиков, методы градуировки, способы формирования отчетов, как в газовой хроматографии и ВЭЖХ. По аналогии с ВЭЖХ большинство детекторов в капиллярном электрофорезе являются концентрационными, для которых высота или площадь пика прямо пропорциональны концентрации вещества, образующего пик.
Качественный анализ. Характеристики миграции/удерживания
Качественный анализ обычно состоит в сравнении времен миграции (в случае капиллярного зонного электрофореза) или времен удерживания (в случае мицеллярной электрокинетической хроматографии), полученных для стандарта и пробы, измеренных в одинаковых условиях. Если эти времена совпадают с заданной точностью (обычно окно идентификации не превышает 5 %), то считают, что искомое вещество в пробе найдено и переходят к количественному анализу. Тем не менее, такой способ идентификации вещества не всегда надежен, особенно в случае анализа проб со сложной матрицей.
Несмотря на высокую разделительную способность капиллярного электрофореза, качественный анализ близкорасположенных пиков может вызывать некоторые трудности. В этом случае можно рекомендовать использование метода добавок. В пробу, для которой затруднена идентификация анализируемого вещества, вносят это вещество и проводят повторный анализ. Если на электрофореграмме появляется новый пик, это означает, что анализируемый компонент ранее в пробе отсутствовал. Если же один из бывших пиков увеличился по высоте (площади), то можно утверждать, что это и есть анализируемый компонент. Величину добавки обычно выбирают так, чтобы высота (площадь) интересующего нас пика увеличилась не более чем в 2—3 раза.
Зачастую приходится сталкиваться с ситуацией, когда время миграции компонента не стабильно от анализа к анализу, что связано, в том числе, с нестабильностью электроосмотического потока. Причин этому несколько, от недостаточно кондиционного состояния капилляра, использования модификации внутренней поверхности капилляра или введения добавок в состав буферного электролита до температурных эффектов и влияния матричных и сопутствующих компонентов. Использование в таких ситуациях маркера ЭОП (например, ацетона) как в растворе стандарта, так и в пробе, позволит вычислить исправленные времена миграции, представляющие собой разность времен миграции анализируемого вещества и метки ЭОП.
Еще одним из вариантов повышения достоверности идентификации анализируемого компонента является введение в стандартный раствор и раствор пробы маркера — внутреннего стандарта. Это должно быть вещество, заведомо отсутствующее в анализируемых пробах, но имеющее схожие с определяемым веществом физико-химические свойства. Для стандарта и пробы вычисляют относительные времена миграции (можно арифметически поделить время миграции компонента на время миграции ЭОП и, наоборот, но для пробы и для стандарта это должно быть сделано одинаково) и находят в пробе близкие по численному значению результаты.
Наиболее полную и достоверную идентификацию вещества на сегодняшний день можно получить при использовании диодно-матричного детектора, который по результату одного анализа может предоставить информацию:
по сопоставлению времени миграции вещества и его спектра в пробе и стандартном растворе (при этом дополнительно будет дана оценка чистоты пика пробы, например, по наложению спектров, снятых в трех точках пика: на обоих склонах и в максимуме);
по отношению откликов пика (например, площади) на двух разных длинах волн, полученных для стандарта и пробы. Для одного и того же вещества на двух разных длинах волн при неизменном времени миграции отношение площадей в стандартном растворе и растворе пробы должно быть постоянным. Длины волн выбирают так, чтобы компонент имел при этом разное поглощение, т. е. высота или площадь пика при двух разных длинах волн были бы различными.
Количественная обработка результатов анализа
В основе количественного анализа лежит прямо пропорциональная зависимость высоты (площади) пика от концентрации вещества при использовании концентрационных детекторов, какими являются, например, фотометрические и флуориметрические детекторы.
Суть количественного определения сводится к следующему: сначала выбирают метод градуировки (внешнего стандарта (абсолютной градуировки), внутреннего стандарта, метод добавок и т. д.); определяют какую величину отклика детектора — высоту пика или площадь пика — будут использовать; затем анализируют стандартные растворы с известными концентрациями веществ и для каждого компонента строят градуировочную зависимость отклика детектора от концентрации вещества; после чего анализируют пробу неизвестного состава и по градуировочному графику находят концентрацию определяемых веществ.
Основным методом градуировки является метод внешнего стандарта (абсолютной градуировки), для которого необходимо иметь ГСО или химически чистые стандарты всех определяемых компонентов. Градуировка может быть одноточечной и многоточечной. Одноточечная означает, что для градуировки компонента используется только один градуировочный раствор, зависимость носит строго линейный характер и, как правило, выходит из начала координат. Это частный случай многоточечной градуировки, для построения которой анализируют несколько специально подобранных по концентрациям градуировочных растворов, после чего с помощью метода наименьших квадратов рассчитывают коэффициенты прямой, наилучшим образом описывающей экспериментальные данные. Правильное и тщательное проведение градуировки является необходимым условием точности получаемых количественных результатов анализа.
Основными областями применения метода КЭ являются:
· Анализ объектов окружающей среды: природные, питьевые, сточные воды и почвы (анионы, катионы, гербициды).
· Контроль качества пищевой продукции и продовольственного сырья: (неорганические катионы и анионы, консерванты, органические кислоты, подсластители, синтетические красители, антиоксиданты, аминокислоты, витамины, углеводы, белки).
· Анализ показателей качества кормов, комбикормов и сырья для их производства: (аминокислоты, белки, витамины).
· Фармация: технологический контроль и анализ готовых лекарственных форм, разделение оптических изомеров.
· Клиническая биохимия: определение неорганических катионов и анионов, аминокислот, белков в биологических жидкостях, определение фармакокинетики лекарственных препаратов.
· Криминалистическая экспертиза: обнаружение остаточных количеств взрывчатых веществ, анализ наркотических средств.
· Химическая промышленность: технологический контроль, определение состава сырья и полупродуктов.
|
|