Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Индукция сепсиса и септического шока
|
|
При септическом шоке гиперцитокинемия повышает активность инду-цибельной синтетазы оксида азота в эндотелиальных и других клетках. В результате снижается сопротивление резистивных сосудов и венул. Снижение тонуса данных микрососудов уменьшает общее периферическое сосудистое сопротивление. Это снижает уровень возбуждения рецепторов транспортно-демпферного отдела системного кровообращения. Активность вагальных кардиальных нейронов падает, и вследствие тахикардии минутный объем кровообращения растет.
Несмотря на рост минутного объема кровообращения, часть клеток организма при септическом шоке страдает от ишемии, обусловленной расстройствами периферического кровообращения. Расстройства периферического кровообращения при сепсисе и септическом шоке — это следствия системной активации эндоте-лиоцитов, полиморфоядерных нейтрофилов и мононуклеарных фагоцитов. В активированном состоянии данные клетки осуществляют адгезию и экзоцитоз, что разрушает стенки микрососудов. Ишемия при сепсисе отчасти возникает вследствие спазма резистивных сосудов и прекапиллярных сфинктеров, который связан с дефицитом активности конституционной синтетазы оксида азота эндоте-лиоцитов и других клеток.
Реакция системного кровообращения на возникновение воспалительного очага определенной распространенности направлена на уничтожение и элиминацию источников чужеродных антигенов, в том числе и своих некробиотически измененных тканей. При этом рост минутного объема кровообращения (МОК) отчасти представляет следствие выхода в кровь и супрасегментарного действия первичных провоспалительных цитокинов (фактора некроза опухолей-альфа и др.), которое повышает МОК. Рост МОК увеличивает доставку лейкоцитов в очаг воспаления. Кроме роста МОК системную воспалительную реакцию и сепсис характеризует снижение общего периферического сосудистого сопротивления посредством дилатации сосудов сопротивления на периферии. Это повышает доставку лейкоцитов в капилляры. Если в физиологических условиях нейтрофилы незатрудненно минуют артериолы, капилляры и венулы, то при гиперцитокинемии они задерживаются эндотелиоцитами венул. Дело в том, что гиперцитокинемия, повышая экспрессию адгезивных молекул на поверхности как эндотелиоцитов, так и нейтрофилов, вызывает адгезию полиморфонуклеаров к эндотелиальным клеткам второго типа, выстилающим стенку венул. Адгезия выступает начальным этапом патогенного воспаления, не имеющего защитного значения. До стабильной адгезии посредством одновременной экспрессии и соединения друг с другом адгезивных молекул эндотелиоцитов и полиморфноядерных лейкоцитов происходит роллинг (катание) нейтрофилов по поверхности эндотелия. Роллинг и адгезия — необходимые этапы трансформации нейтрофилов в клетки, осуществляющие воспаление и способные к экзофагоцитозу. Это те этапы воспаления, после реализации которых почти полностью развертывается последовательность причин и следствий, составляющих данную защитно-патогенную реакцию.
Воспаление данного генеза имеет сугубо патологический характер, происходит во всех органах и тканях, повреждая элементы исполнительных аппаратов. Критическое падение числа структурно-функциональных элементов большинства органов-эффекторов составляет основное звено патогенеза так называемой множественной системной недостаточности.
Адгезия приводит к обтурации венул, что повышает гидростатическое давление в капиллярах и массу ультрафильтрата, поступающую в интерстиций.
Согласно традиционным и верным представлениям сепсис, и системную воспалительную реакцию вызывает патогенное действие грамотрицательных микроорганизмов.
В индукции системной патологической реакции на инвазию во внутреннюю среду и кровь грамотрицательных микроорганизмов определяющую роль играют:
♦ Эндотоксин (липид А, липополисахарид, ЛПС). Этот термостабильный липополисахарид составляет наружное покрытие грамотрицательных бактерий. Эндотоксин, воздействуя на нейтрофилы, вызывает высвобождение полиморфноядерными лейкоцитами эндогенных пирогенов.
♦ ЛПС-связывающий белок (ЛПССБ), следы которого определяют в плазме в физиологических условиях. Данный протеин образует с эндотоксином молекулярный комплекс, циркулирующий с кровью.
♦ Рецептор клеточной поверхности мононуклеарных фагоцитов и эндотелиальных клеток. Его специфическим элементом является молекулярный комплекс, состоящий из ЛПС и ЛПССБ (ЛПС-ЛПССБ). Рецептор составляют TL-рецептор и поверхностный маркер лейкоцитов CD 14.
В настоящее время растет частота сепсиса, обусловленного инвазией во внутреннюю среду грамположительных бактерий. Индуцирование сепсиса грамположительными бактериями обычно не связано с высвобождением ими эндотоксина. Известно, что предшественники пептидогликана и другие компоненты стенок грамположительных бактерий вызывают высвобождение клетками систем иммунитета фактора некроза опухолей-альфа и интерлейкина-1. Пептидогликан и другие компоненты стенок грамположительных бактерий активируют систему комплемента по альтернативному пути. Активация системы комплемента на уровне всего организма вызывает системное патогенное воспаление и вносит свой вклад в эндотоксикоз при сепсисе и системной воспалительной реакции.
Большинство из опытных клиницистов с легкостью распознают состояние септического шока (СШ). Если тех же врачей-исследователей попросить дать определение данного патологического состояния, то будет дано множество разных дефиниций, во многом противоречащих друг другу. Дело в том, что патогенез септического шока остается во многом неясным. Несмотря на многочисленные исследования патогенеза септического шока, антибиотики остаются средствами, действие которых составляет главный этиопатогенетический элемент терапии при СШ. При этом летальность среди пациентов в септическом шоке составляет 40-60%. Исследования, направленные на ослабление действия некоторых из медиаторов септического шока, не привели к разработке эффективной терапии. В настоящее время остается неясным, должна ли система терапии быть ориентированной на блокаду действия какого-либо одного из ведущих звеньев патогенеза септического шока, или у каждого из пациентов лечение должно быть строго индивидуализированным.
Септический шок — это совокупность расстройств функциональных систем, при которых артериальная гипотензия и недостаточная объемная скорость тока крови на периферии не подвергаются обратному развитию под действием внутривенных инфузий тех или иных плазмозамещающих средств. Это результат не ограниченного системной регуляцией действия некоторых из механизмов врожденных иммунных реакций. Врожденные иммунные реакции обладают собственными бактерицидными эффектами, а также готовят и вызывают приобретенные клеточные и гуморальные иммунные реакции.
Реакции врожденного иммунитета во многом вызываются взаимодействием лигандов патогенов с гуморальными и клеточными рецепторами организма. Одни из таких рецепторов — это TL-рецепторы (англ. toll-like, со свойствами барьера, «набата», «передового охранения»). В настоящее время известно более десяти разновидностей TL-рецепторов млекопитающих. Соединение лиганда бактериального происхождения с TL-рецептором запускает комплекс клеточных реакций. В результате этих реакций оказывается бактерицидное действие, индуцируется воспаление и происходит подготовка к специфической иммунной реакции. При избыточности комплексной реакции систем врожденного иммунитета возникает септический шок.
Существует несколько уровней, на которых представляется возможным заблокировать патологическую реакцию систем врожденного иммунитета, вызывающую септический шок. Первый из них — это уровень взаимодействия экзогенных бактериальных лиганд с гуморальными и клеточными рецепторами систем врожденного иммунитета. Ранее считалось, что септический шок всегда вызывается эндотоксином (липополисахаридом бактериального происхождения), который высвобождается грамотрицательными бактериями. Теперь общепризнанным является тот факт, что менее чем в 50% случаев септический шок вызывается грамположительными возбудителями. Грамположительные патогены, высвобождают компоненты своей стенки, аналогичные по структуре эндотоксину. Эти компоненты способны вызывать септический шок, вступая во взаимодействие с клеточными рецепторами (рецепторами наружной поверхности мононуклеарных фагоцитов). Следует заметить, что при обследовании больного весьма трудно определить механизм индукции септического шока.
Возникновение септического шока своим необходимым условием имеет гиперцитокинемию, то есть рост концентрации в циркулирующей крови первичных провоспалительных цитокинов. В этой связи были предложены различные способы блокады действия первичных провоспалительных цитокинов (моноклональные антитела к фактору некроза опухолей-альфа и др.), которые не снизили летальности при септическом шоке. Дело в том, что воздействие оказывалось лишь на один элемент иммунопатологической реакции. Избрать мишенью терапии один противоспалительный цитокин — это означает влиять лишь на одно из многих одновременных и одинаковых по значению звеньев патогенеза септического шока.
Итак, можно считать, что в настоящее время известен ряд в эволюционном отношении древних лигандов, принадлежащих грамотрицательным и грамположительным бактериям, а также микобактериям и грибковым возбудителям. Данные экзогенные лиганды способны взаимодействовать с небольшим числом гуморальных и клеточных рецепторов, вызывая сепсис и септический шок. В этой связи нельзя исключить, что в будущем патологическую реакцию систем врожденного иммунитета можно будет оптимально блокировать, воздействуя на гуморальные и клеточные рецепторы лиганд бактерий, ответственных за возникновение септического шока.
Для распознавания своих лиганд TL-рецепторам необходимы вспомогательные молекулы. Очевидно, что предстоит еще выявить тот гуморальный рецептор (плазменный белок), который связывается с элементами наружной мембраны грамположительных бактерий.
Прежде чем молекулярный комплекс компонента бактериальной стенки и гуморального рецептора свяжется с TL-рецептором, он соединяется с CD 14. В результате TL-рецептор активируется, то есть начинается передача сигнала генам клетки о начале экспрессии первичных провоспалительных цитокинов и бактерицидных агентов. Существует принципиальная возможность предотвратить индукцию септического шока воздействием на CD14. Кроме того, представляется теоретически возможным блокировать патогенез септического шока в зародыше, блокируя TL-рецепторы, а также передачу генерируемого ими сигнала на пострецепторном внутриклеточном уровне.
4.Этиология и патогенез
Септический шок — это наиболее частая причина летальных исходов в хирургических стационарах и отделениях интенсивной терапии. Термины «сепсис», «тяжелый сепсис», «септический шок» соответствуют разным степеням тяжести патологической реакции организма и системы иммунитета на инфекцию. В основном сепсис как синдром характеризуют признаки инфекции и воспаления. При тяжелом сепсисе в различных органах падает объемная скорость тока крови, что вызывает сочетанные расстройства функциональных систем (множественную системную недостаточность). Возникновение септического шока знаменует устойчивая артериальная гипотензия. Смертность при сепсисе — 16%, а при септическом шоке — 40-60%.
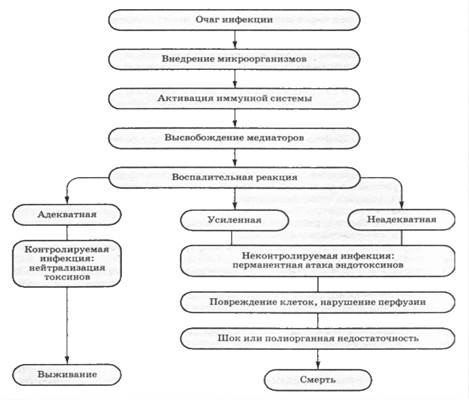
Бактериальная инфекция — это наиболее частая, причина септического шока. При сепсисе первичные очаги инфекции чаще локализованы в легких, органах живота, брюшине, а также в мочевыводящих путях. Бактериемию выявляют у 40-60% больных в состоянии септического шока. У 10-30% больных в состоянии септического шока невозможно выделить культуру бактерий, действие которых вызывает септический шок. Можно предположить, что септический шок без бактериемии — это результат патологической иммунной реакции в ответ на стимуляцию антигенами бактериального происхождения. По-видимому, данная реакция сохраняется после элиминации из организма патогенных бактерий действием антибиотиков и других элементов терапии, то есть происходит ее эндогенизация. В основе эндогенизации сепсиса могут лежать многочисленные, усиливающие друг друга и реализуемые через выброс и действие цитокинов, взаимодействия клеток и молекул систем врожденного иммунитета и, соответственно, иммуно-компетентных клеток. Раньше тяжелый сепсис и септический шок связывали исключительно с грамотрицательными аэробными бациллами. В настоящее время частота грамположительной инфекции как причины сепсиса равна частоте сепсиса, обусловленного инвазией во внутреннюю среду грамотрицательных микроорганизмов. Это произошло из-за широкого использования внутрисосудистых катетеров, других устройств, так или иначе находящихся во внутренней среде, а также из-за роста частоты пневмоний. Грибковые, вирусные и протозойные инфекции также могут быть причинами септического шока.
Системная воспалительная реакция индуцируется высвобождением из очага воспаления самих патогенных бактерий, их токсинов, а также цитокинов со свойствами медиаторов воспаления. В наибольшей степени как индуктор системной воспалительной реакции изучен эндотоксин грамотрицательных аэробных бацилл. Кроме того, известны другие бактериальные продукты (токсины), которые могут вызвать массивный выброс клетками систем врожденного иммунитета медиаторов воспаления. К таким бактериальным продуктам относят формиловые пептиды, экзотоксины, энтеротоксины, гемолизины-протеогликаны, а также липотейхоевую кислоту, которую образуют грамположительные микроорганизмы.
Бактериальные токсины стимулируют высвобождение мононуклеарными фагоцитами цитокинов со свойствами медиаторов воспаления, которые сначала вызывают, а затем усиливают системную воспалительную реакцию. Токсины связываются со своими клеточными рецепторами, активируя регуляторные белки. В частности, таким образом активируется транскрипционный фактор NF-kB. В активированном состоянии NF-kB усиливает экспрессию генов цитокинов со свойствами медиаторов воспаления.
Активация NF-kB прежде всего повышает образование мононуклеарными фагоцитами фактора некроза опухолей-альфа и интерлейкина-1. Данные цитокины называют первичными провоспалительными. Фактор некроза опухолей-альфа и интерлейкин-1 стимулируют высвобождение мононуклеарными фагоцитами, а также иммунокомпетентными клетками интерлейкинов 6 и 8 и других медиаторов воспалительной реакции: тромбоксанов, лейкотриенов, фактора активации тромбоцитов, простагландинов и активированных фракций системы комплемента.
Полагают, что оксид азота служит главным медиатором системного расширения сосудов, падения общего периферического сосудистого сопротивления и артериальной гипотензии у больных в состоянии септического шока.
Индуцируемая (индуцибельная) форма синтетазы оксида азота экспрессиру-ется и высвобождается эндотелиальными и другими клетками только при определенных условиях. К одному из таких условий относится действие на эндотелиоциты первичных провоспалительных цитокинов. Вызывая экспрессию индуцируемой формы синтетазы в эндотелиальных, гладкомышечных клетках сосудистой стенки и мононуклеарных фагоцитах, первичные провоспалительные цитокины повышают высвобождение оксида азота на системном уровне. Усиление действия оксида азота на системном уровне снижает общее периферическое сосудистое сопротивление и вызывает артериальную гипотензию. При этом оксид азота служит субстратом образования пероксинитрита, то есть продукта реакции NO со свободными кислородными радикалами, который обладает прямым цитотоксическим действием. Этим не исчерпывается роль оксида азота в патогенезе септического шока. Он оказывает отрицательное инотропное действие на сердце и повышает проницаемость стенки микрососудов.
Угнетение сократимости сердца при септическом шоке происходит также вследствие отрицательного инотропного действия фактора некроза опухолей-альфа.
Действие фактора некроза опухолей-альфа вызывает отек митохондрий и повреждает митохондриальные цепи дыхательных ферментов. В результате в клетке возникает дефицит свободной энергии, и наступает клеточная гибель, обусловленная гипоэргозом. Известно, что митохондрии — это главный источник свободных кислородных радикалов, высвобождаемых в цитозоль клетки. Действие марганцевой супероксиддисмутазы инактивирует О2-, который высвобождается цепью дыхательных ферментов. При этом антиоксидант предотвращает апоптоз, который вызывается фактором некроза опухолей-альфа. Это заставляет считать механизм апоптоза под действием фактора некроза опухолей-альфа связанным с высвобождением митохондриями свободных кислородных радикалов. Образование свободных кислородных радикалов митохондриями растет под действием фактора некроза опухолей-альфа. При этом свободные кислородные радикалы, высвобождаемые митохондриями, повреждают цепи их дыхательных ферментов. Определенная активность цепей дыхательных ферментов митохондрий — необходимое условие апоптотического действия фактора некроза опухолей-альфа. В эксперименте было показано, что угнетение тканевого дыхания в митохондриях вызывает резистентность клетки по отношению к апоптотическому действию фактора некроза опухолей-альфа. Можно предположить, что клетки с особо высоким содержанием митохондрий и повышенной активностью цепей дыхательных ферментов обладают особо выраженной чувствительностью к действию фактора некроза опухолей-альфа, повреждающему цепи дыхательных ферментов митохондрий и вызывающему гипоэргоз клетки. Такими клетками являются кардиомиоциты. Поэтому действие фактора является особо выраженным на уровне миокарда, сократимость которого падает при шоке. При этом системное повреждающее действие фактора некроза опухолей-альфа на митохондрии может лежать в основе тканевой гипоксии при септическом шоке.
В ответ на действие флогогенов, высвобождаемых при септическом шоке, растет экспрессия адгезивных молекул на поверхности эндотелиоцитов и нейтрофилов. В частности, на поверхности нейтрофилов появляется интегриновый комплекс (CD11/CD18), что происходит одновременно с появлением на поверхности эндо-телиальной клетки межклеточных адгезивных молекул, комплементарных по отношению к интегриновому комплексу. Экспрессия на поверхности нейтрофилов интегринового комплекса — это одно из следствий активации данных клеток. Расстройства периферического кровообращения при септическом шоке, адгезия активированных полиморфоядерных лейкоцитов к активированным эндотелиоцитам — все это ведет к выходу нейтрофилов в интерстиций и воспалительной альтерации клеток и тканей. Одновременно эндотоксин, фактор некроза опухолей-альфа, а также интерлейкин-1 повышают образование и высвобождение эндотелиальными клетками тканевого фактора свертывания. В результате активируются механизмы внешнего гемостаза, что вызывает отложение фибрина и диссеминированное внутрисосудистое свертывание.
При септическом шоке рост экспрессии и высвобождения провоспалительных цитокинов вызывает патологическую реакцию выброса в интерстиций и кровь эндогенных иммунодепрессантов. Это обуславливает фазу иммунодепрессии септического шока.
Индукторами иммунодепрессии при септическом шоке являются: 1) кортизол и эндогенные катехоламины; 2) интерлейкины 10 и 4; 3) простагландин Е2; 4) растворимые рецепторы фактора некроза опухолей; 5) эндогенный антагонист рецептора интерлейкина-1 и др. Растворимые рецепторы фактора связывают его в крови и межклеточных пространствах. При иммунодепрессии падает содержание антигенов тканевой совместимости второго типа на поверхности мононуклеар-ных фагоцитов. Без таких антигенов на своей поверхности мононуклеары не могут действовать в качестве антиген-презентирующих клеток. Одновременно угнетается нормальная реакция мононуклеаров на действие медиаторов воспаления. Все это может служить причиной нозокомиальных инфекций и смерти.
Артериальная гипотензия при септическом шоке в основном представляет собой следствие снижения общего периферического сосудистого сопротивления. Гиперцитокинемия и рост концентрации в крови оксида азота при септическом шоке обуславливает расширение артериол. При этом посредством тахикардии компенсаторно растет минутный объем кровообращения. Артериальная гипотензия при септическом шоке возникает, несмотря на компенсаторный рост минутного объема кровообращения. Общее легочное сосудистое сопротивление при септическом шоке растет, что можно отчасти связать с адгезией активированных нейтрофилов к активированным эндотелиоцитам легочных микрососудов.
При септическом шоке выявляют следующие признаки юкстакапиллярного шунтирования крови:
1) лактатный ацидоз;
2) снижение артериовенозного различия по кислороду, то есть различия в содержании кислорода между артериальной и венозной кровью.
При септическом шоке расширены емкостные сосуды, что приводит к общей венозной гиперемии. Расширение артериол и вен выражено при септическом шоке по-разному в разных областях. Это определяет патологическую вариабельность пре- и посткапиллярных сосудистых сопротивлений. Патологическая вариабельность вызывает аномальное перераспределение минутного объема кровообращения и объема циркулирующей крови. Дилатация сосудов при септическом шоке наиболее выражена в очаге воспаления. Расширение сосудов при септическом шоке связано с ростом содержания в крови эндогенных вазодилататоров и снижением чувствительности альфа-адренорецепторов сосудистой стенки к эндогенным катехоламинам.
Выделяют следующие основные звенья патогенеза расстройств периферического кровообращения при септическом шоке:
1)рост проницаемости стенки микрососудов;
2) рост сопротивления микрососудов, который усиливается клеточной адгезией в их просвете;
3) низкая реакция микрососудов на вазодилатирующие влияния;
4) артериоло-венулярное шунтирование;
5) падение текучести крови.
В эксперименте было показано, что общая площадь поперечного сечения капилляров у экспериментальных животных в состоянии септического шока снижается. Это следствие патогенных межклеточных взаимодействий с участием эндотелиальных клеток. Снижение общего просвета капилляров у больных в состоянии септического шока проявляется угнетением реактивной гиперемии. Реактивную гиперемию угнетают расстройства местной регуляции тока крови по микрососудам и падение способности клеток крови проходить по капиллярам. В частности, такую способность снижает появление на поверхности нейтрофилов и моноцитов адгезивных молекул. Кроме того, данная способность падает вследствие снижения деформируемости нейтрофилов и эритроцитов. Известно, что при септическом шоке снижается активность конституционной (постоянно присущей клеточному фенотипу) синтетазы оксида азота. Действие конституционной синтетазы повышает ток крови на периферии. Падение активности данного фермента снижает ток крови на периферии, что угнетает реактивную гиперемию. У больных в состоянии септического шока выявляют отек эндотелиоцитов, депозиты фибрина в микрососудах и межклеточных пространствах, увеличение адгезивной способности нейтрофилов и эндотелиальных клеток, а также образование агрегатов из нейтрофилов, тромбоцитов и эритроцитов в венулах, артериолах и капиллярах. В некоторых случаях возникает открытие артериоло-венулярных анастомозов как причина юкстакапиллярного шунтирования.
Гиповолемия — это один из факторов артериальной гипотензии при септическом шоке. Выделяют следующие причины гиповолемии (падения преднагрузки сердца) у больных в состоянии септического шока: 1) дилатация емкостных сосудов; 2) потеря жидкой части плазмы крови в интерстиций из-за патологического роста проницаемости капилляров. Падение преднагрузки сердца и общего периферического сосудистого сопротивления — это еще не все причины артериальной гипотензии при септическом шоке. Ее также обуславливает отрицательное действие на сердце медиаторов септического шока. И левый, и правый желудочки сердца при септическом шоке последовательно проходят стадии ригидности (недостаточности диастолической функции) и дилатации (недостаточности систолической функции). Ригидность и дилатация не связаны с падением тока крови по венечным артериям и ростом потребности кардиомиоцитов в кислороде. Насосную функцию сердца при септическом шоке угнетают фактор некроза опухолей-альфа, а также интерлейкин-1. Угнетение насосной функции сердца при септическом шоке отчасти связано с легочной артериальной гипертензией и падением чувствительности бета-адренорецепторов сердца.
Можно считать, что у большинства больных в состоянии септического шока падение потребления кислорода организмом преимущественно обусловлено первичными нарушениями тканевого дыхания. При кардиогенном шоке лактатный метаболический ацидоз вызывается тяжелой циркуляторной гипоксией. При этом напряжение кислорода в смешанной венозной крови находится на уровне ниже 30 мм рт. ст. При септическом шоке умеренный лактатный ацидоз развивается при нормальном напряжении кислорода в смешанной венозной крови. Лактатный ацидоз при септическом шоке считают следствием снижения активности пируватдегидрогеназы и вторичной аккумуляции лактата, а не падения тока крови на периферии. В случае септического шока причины падения улавливания клеткой свободной энергии при аэробном биологическом окислении — это цито-токсические эффекты (прямые или опосредованные) эндотоксина, оксида азота, фактора некроза опухолей-альфа. Патогенез септического шока во многом складывается из расстройств биологического окисления и определяется гипоэргозом клетки как следствием тканевой гипоксии, развившейся под действием эндотоксемии.
Расстройства периферического кровообращения при сепсисе носят системный характер и развиваются при артериальной нормотензии, которую поддерживает рост минутного объема кровообращения. Системные нарушения микроциркуляции проявляют себя снижением рН в слизистой оболочке желудка и падением насыщения кислородом гемоглобина крови в печеночных венах.
Гипоэргоз клеток кишечного барьера, действие иммуносупрессивных звеньев патогенеза септического шока — все это снижает защитный потенциал кишечной стенки, что служит еще одной из причин эндотоксемии при септическом шоке.
|
|