Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Декларация о соответствии оформляется в одном экземпляре и не имеет копии.
|
|
Держателем декларации является изготовитель, поставщик или продавец.
Декларация о соответствии представляет собой документ, в котором производитель (поставщик) декларирует (заявляет), что готовая продукция (серия) ЛС, которую он выпускает в обращение, соответствует стандартам качества, принятым в РФ (ФС, ФСП, НД).
Цель декларирования качества готовой продукции заключается в том, что обращение лекарственных средств на территории Российской Федерации согласно действующему законодательству возможно только после подтверждения соответствия его качества требованиям нормативных документов (ОФС, ФС, ФСП, НД на лекарственные средства зарубежного производства).
Декларацию о соответствии ЛС принимает юридическое или физическое лицо, которое зарегистрировано в качестве индивидуального предпринимателя. Это лицо является изготовителем или продавцом ЛС или выполняет функции иностранного изготовителя на основании договора с ним (в части обеспечения соответствия поставляемой продукции требованиям технических регламентов и ответственности за несоответствие поставляемой продукции требованиям технических регламентов).
Для декларирования соответствия качества продукции требованиям НД разработаны следующие схемы:
- принятие декларации о соответствии на основании собственных доказательств;
- принятие декларации о соответствии на основании собственных доказательств и доказательств, полученных с участием органа по сертификации и (или) аккредитованной испытательной лаборатории (центра) (третья сторона).
Для лекарственных средств согласно Федеральному закону от 27.12.02. № 184-ФЗ «О техническом регулировании» Минпромэнерго РФ приказом от 22.03.06 № 54 и Постановлением Правительства РФ от 07.07.99 № 766 утверждена схема декларирования соответствия ЛС требованиям технических регламентов с участием третьей стороны.
Согласно этой схеме в декларировании участвуют (рис. 1):
- декларант (предприятие-изготовитель; поставщик);
- аккредитованный испытательный центр (испытательная лаборатория);
- аккредитованный орган по сертификации (федеральный; региональный).

Рисунок 1 – Схема декларирования качества готовой продукции серии лекарственного средства предприятием-изготовителем
Процесс декларирования заключается в том, что путем принятия декларации декларант (предприятие-изготовитель; поставщик) заявляет, что продукт, который он выпускает в обращение, соответствует стандартам качества, утвержденным в РФ. При этом декларант предъявляет необходимые собственные доказательства соответствия продукции требованиям НД и результаты независимой экспертизы качества каждой серии готового продукта, выполненные аккредитованной испытательной лабораторией (центром) (третья сторона).
Декларация о соответствии принимается в отношении каждой серии (партии) лекарственного средства, выпускаемого в обращение, на срок, установленный изготовителем (продавцом) ЛС, но не более установленного срока годности ЛС.
Для проведения независимой экспертизы качества каждой серии готового продукта декларант самостоятельно отбирает образцы или может поручить отбор образцов на договорной основе испытательной лаборатории или органу по сертификации. Обязательное условие при отборе проб – неукоснительное соблюдение законодательных актов в соответствии с нормативными документами, составление акта отбора образцов и предоставление его в испытательную лабораторию.
При подтверждении соответствия качества готового продукта (серии) лекарственного средства требованиям НД в форме декларирования декларанты (предприятие-изготовитель; продавец) могут самостоятельно выбирать испытательную лабораторию и орган по сертификации, который регистрирует декларацию о соответствии, для чего заключает с ними договоры.
Перечень аккредитованных испытательных лабораторий приведен в письме Росздравнадзора от 23.03.07 № 01И-197/07. При изменении реестра аккредитованных испытательных лабораторий, Росздравнадзор доводит данную информацию до сведения субъектов обращения ЛС публикацией на сайте (www.roszdravnadzor.ru).
Декларация о соответствии может быть направлена на регистрацию и зарегистрирована только в одном органе по сертификации по выбору изготовителя (продавца).
Аккредитованный испытательный центр (испытательная лаборатория) проводит независимую экспертизу и определяет соответствие качества ЛС установленным требованиям и заявленному в декларации.
Орган по сертификации, в установленном порядке аккредитованный Ростехрегулированием на данный вид деятельности (перечень органов по сертификации приведен в письме Росздравнадзора от 13.12.06 № 01И-946/06), в течение 7 дней проверяет декларацию и всю представленную документацию по следующим параметрам:
- полнота выполнения анализа согласно показателям качества в нормативном документе;
- правильность оформления и подлинность документов;
- компетентность органов, выдавших соответствующие документы.
При положительном решении Орган по сертификации, аккредитованный в установленном порядке, регистрирует ДЕКЛАРАЦИЮ СООТВЕТСТВИЯ и включает в реестр зарегистрированных деклараций.
В соответствии с нормативными правовыми актами в каждой организации-производителе (поставщике) ЛС должно быть назначено лицо, уполномоченное на принятие (подписание) декларации.
В пакете документов для декларации соответствия готового продукта (серии) лекарственных средств в органах по сертификации должны быть представлены следующие документы:
- Декларация о соответствии (приложение 4);
- паспорт производителя;
- протоколы входного контроля на сырье и материалы, используемые в производстве (приложение 1);
- копия документа о внесении юридического лица в единый государственный реестр;
- копия регистрационного удостоверения на лекарственное средство;
- копия лицензии на производство лекарственного средства;
- копия протокола испытаний, проведенного в испытательной лаборатории (независимая экспертиза) (приложение 3).
В качестве собственных доказательств соответствия готовой продукции требованиям НД производитель продукции представляет следующие документы:
- Паспорт (протокол анализа) производителя для отечественных лекарственных средств (приложение 1);
- Сертификат качества (анализа) фирмы для зарубежных лекарственных средств, выданный на соответствие показателям качества, установленным в НД на данное лекарственное средство (ЛС);
- Документ, подтверждающий происхождение ЛС;
- Протоколы входного контроля (испытаний) на сырье, полупродукты, субстанции и материалы, используемые в производстве ЛС (приложение 2);
- Документы, подтверждающие происхождение сырья, используемого в производстве данного ЛС.
Ответственность за качество ЛС несут производители ЛС или лица, выполняющие функции иностранного изготовителя на основании договора с ним в части обеспечения соответствия поставляемой продукции требованиям технических регламентов и в части ответственности за несоответствие поставляемой продукции требованиям технических регламентов.
Каждый участник фармацевтического рынка должен внедрить систему обеспечения качества. Каждый субъект обращения ЛС (производитель, поставщик, аптека) на своем участке несет ответственность за качество реализуемых лекарственных средств.
Участники фармацевтического рынка могут идентифицировать сведения деклараций о соответствии ЛС требованиям НД, содержащиеся в товарно-сопроводительных документах, по базе зарегистрированных деклараций, размещенной на сайте ФГУ «Центр экспертизы и контроля качества медицинской продукции» Росздравнадзора www. fgusertif.ru.
В дальнейшем планируется создание на сайте Росздравнадзора раздела по данным базы зарегистрированных деклараций о соответствии, с помощью которого субъекты обращения лекарственных средств и население смогут проверить факт регистрации декларации любого лекарственного средства, находящегося в обращении.
Для успешного функционирования системы декларирования качества ЛС на фармацевтических предприятиях создана Система обеспечения качества лекарственных средств. Она включает Отдел Обеспечения Качества (ООК), Отдел Контроля Качества (ОКК) и, в пределах юрисдикции, производственные подразделения и складскую зону (рис. 2).
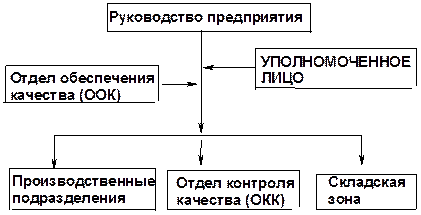
Рисунок 2 – Организационная структура системы обеспечения качества
лекарственных средств на фармацевтическом предприятии
Согласно схеме (рис. 2)заместителем руководителя предприятия по качествуявляется УПОЛНОМОЧЕННОЕ ЛИЦО.
УПОЛНОМОЧЕННОЕ ЛИЦО на основании изучения материалов «Досье на серию» принимает решение о выпуске в обращение готовой серии лекарственного средства. Материалы Досье должны сформировать у уполномоченного лица уверенность в том, что каждая серия продукции произведена и проверена в соответствии с установленными требованиями. При наличии уверенности в качестве продукции УПОЛНОМОЧЕННОЕ ЛИЦО принимает решение на выпуск готовой продукции в обращение, дает письменное разрешение на выпуск серии лекарственного средства и ставит свою подпись. Подпись УПОЛНОМОЧЕННОГО ЛИЦА на разрешении является гарантией соответствия качества данной серии требованиям регистрационного досье.
Функциями Отдела Обеспечения Качества (ООК) являются:
- создание единой системы документации на производстве, разработка и распределение контролируемой документации;
- контроль над распространением, обращением и своевременным изъятием из оборота контролируемой документации;
- разработка системы идентификации и изменения статуса сырья, материалов, полупродуктов, готовой продукции, контрольных процедур, оборудования, производственных помещений и разработка системы контроля изменений;
- организация аудита поставщиков сырья и материалов, разработка системы отгрузки ЛС потребителю, работы с рекламациями, отзыва ЛС с рынка и работы с возвращенной продукцией;
- разработка порядка проведения валидации и ревалидации, внутренних проверок, контроля выполнения корректирующих действий и расследования несоответствий;
- обеспечение Уполномоченного лица информацией, необходимой для принятия решения о выпуске серии в обращение.
Отдел Контроля Качества (ОКК) является для ООК ближайшим и основным уровнем подчинения в сфере оперативного (точечного) контроля на этапах входного и операционного контроля, а также контроля готовой продукции.
ОКК осуществляет мониторинг всех количественно измеримых параметров сырья и материалов, полупродуктов и готового продукта, а также персонала, оборудования, помещений и окружающей среды с целью отделения (изъятия из процесса) того, что не будет признано соответствующим спецификациям и другим нормативным документам.
ОКК разрабатывает, аттестует (валидирует), внедряет инструкции и методики по контролю качества, хранению контрольных образцов материалов и продукции; контролирует правильность маркировки упаковок с материалами и продукцией; обеспечивает контроль стабильности продукции; участвует в расследовании рекламаций, связанных с качеством продукции и т.д.
Контроль качества, возглавляемый и осуществляющий ОКК, представляет собой совокупность следующих основных процессов, выполняемых на пути следования продукции от сырья и материалов через полупродукты до готового продукта:
- входной контроль;
- операционный (постадийный) контроль (контроль производства);
- контроль персонала;
- контроль окружающей среды;
- контроль качества продукции;
- контроль корректирующих действий;
- контроль при рекламациях.
К числу главных документов системы обеспечения качества относятся «Информация о предприятии», «Руководство по качеству». Кроме того, в систему обеспечения качества входят следующие рабочие документы:
- стандарты организации (СО);
- документированные или стандартные операционные процедуры (СОПы);
- спецификации;
- рабочие инструкции;
- регистрационная документация (заполняемые формы). К ним относятся аналитические листы, журналы, формы протоколов, акты, отчеты, списки, бланки и др., разработанные на предприятии и используемые с целью протоколирования (регистрации) любых действий в рамках производственных, контрольных, складских, валидационных, логистических и любых иных процедур в процессе создания лекарственного средства.
Контроль качествалекарственных средств на фармацевтическом предприятии реализуется как совокупность следующих основных процессов, лежащих на пути следования продукции от сырья и материалов через полупродукты до готового продукта:
- входной контроль;
- операционный (постадийный) контроль (контроль производства);
- контроль персонала;
- контроль окружающей среды;
- контроль качества продукции;
- контроль корректирующих действий;
- контроль при рекламациях.
Процесс контроля качества лекарственных средств в условиях производственного предприятия можно отразить в виде следующей схемы (рис. 3):
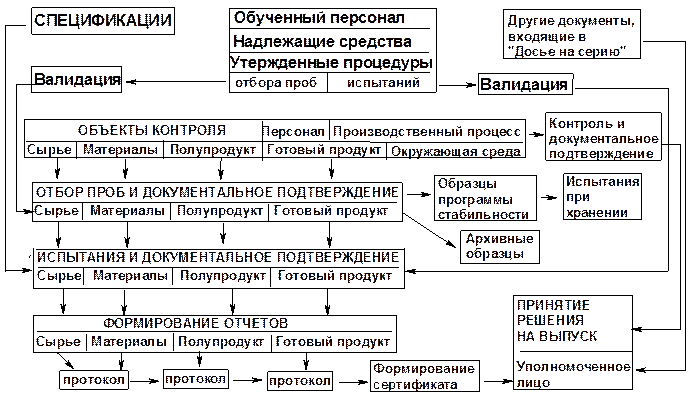
Рисунок 3 – Схема контроля качества ЛС в условиях производственного предприятия.
Уровни контроля, используемые в процессе производства ЛС, подразделяются на оперативный и управленческий.
Оперативный контроль – это первый уровень контроля. Он подразумевает измерение параметров наибольшего количества объектов (проб, отдельных стадий, операций и др.). На этом уровне важнейшее значение приобретают точность, воспроизводимость, скорость, технологичность измерений, их соответствие спецификациям, а также способы их регистрации, анализа, архивирования, хранения, передачи, уничтожения с использованием информационных технологий.
Различают следующие виды оперативного контроля:
- предварительный;
- текущий;
- заключительный.
Каждый из этих видов контроля выполняется выборочно.
Управленческий контроль – это второй уровень контроля. Его задача – проконтролировать качество оценки и качество краткосрочного прогноза.
Управленческий контроль обеспечивает устранение возможных дефектов на промежуточных стадиях изготовления продукта, а не в готовом изделии. Он позволяет предупредить появление дефекта в случае повторения аналогичной ситуации в данной точке производственного процесса.
Управленческий контроль представляет собой высокопрофессиональное оперирование показателями оперативного контроля с целью обеспечения стабильности технологического процесса и гарантированного предупреждения возможности возникновения дефектов.
При управленческом контроле важное значение имеет мониторинг (непрерывное отслеживание (наблюдение) за течением какого-либо процесса путем регистрации характеризующих его параметров) наиболее уязвимых этапов производства и контроля в отношении вероятности появления результатов, выходящих за рамки нормативов или наиболее приближающиеся к их предельно допустимым значениям.
Мониторинг позволяет начальнику ОКК иметь полную информацию о качественных характеристиках выпускаемой продукции на протяжении всего производственного процесса и, при необходимости, своевременно и оперативно вмешаться в производственный процесс для его корректирования.
Управленческий контроль позволяет сформировать базу данных для планирования работы в области обеспечения качества, установление связей между службами контроля качества отдельных производственных подразделений, повышения квалификации персонала.
Управленческий контроль позволяет прогнозировать и контролировать отдаленные последствия принятия того или иного управленческого решения.
Для обеспечения качества выпускаемой продукции предприятие-изготовитель отслеживает качество, начиная с поступления на предприятие сырья и материалов от поставщиков.
С этой целью предприятие осуществляет входной контроль КАЧЕСТВА сырья, материалов, полупродуктов, поступивших от поставщиков для использования в производстве лекарственного средства.
Цель процедур входного контроля – установить единый порядок проведения контроля качества сырья, материалов, полупродуктов с тем, чтобы исключить использование в производстве ЛС некачественных исходных компонентов.
Основные задачи входного контроля:
- контроль качества сырья, материалов, полупродуктов, поступивших от поставщика на предприятие для использования в производстве;
- предупреждение появления брака вследствие поступления на предприятие исходных компонентов ЛС ненадлежащего качества;
- периодический контроль над соблюдением правил и сроков хранения продукции поставщиков;
- своевременная подготовка данных для оформления рекламаций на поступающее сырье в случаях, когда его качество не соответствует требованиям спецификаций;
- периодический контроль над соблюдением правил и сроков хранения продукции поставщиков.
Входной контроль может быть сплошным и выборочным.
Сплошной входной контроль заключается в проверке активных фармацевтических ингредиентов (субстанций) из каждой упаковки одной серии, поступившей на предприятие, на соответствие требованиям НД.
Сплошной входной контроль проводят в случае:
- риска выпуска продукции, не соответствующей требованиям НД, из-за низкого качества поступающего на предприятие сырья и материалов в условиях отсутствия альтернативного поставщика;
- повышенных требований к производимой продукции и применении в производстве контролируемого сырья (например, при производстве стерильных ЛС, предназначенных для введения в спинномозговую жидкость);
- при обнаружении множественных дефектов в ходе выборочного контроля.
Многие фармпредприятия проводят сплошной входной контроль субстанций.
Выборочный входной контроль заключается ввыборочном контроле активных фармацевтических ингредиентов (субстанций) из упаковок одной серии, поступившей на предприятие. Выборочный входной контроль проводят при условии стабильного качества продукции поставщика и гарантированного выпуска качественной собственной продукции.
Входной контроль качества сырья (субстанций, полупродуктов и т.д.) предусматривает проведение двух самостоятельных процедур:
- отбор проб и их доставка в лабораторию отдела контроля качества (ОКК);
- собственно контроль и испытания в ОКК согласно требованиям спецификаций и НД.
При выборочном входном контроле важно отобрать репрезентативную (представительную, т.е. дающую достоверную и объективную информацию о качестве продукции всей серии) выборку при отборе проб поступающего на предприятие исходного сырья для производства лекарственного средства.
В настоящее время для определения представительной пробы используют подход, основанный на стандарте MIL-STD-105D (США). Он позволяет выбрать необходимое число упаковок (m) в зависимости от объема поставки (n) и оценить качество поставленной партии (сырья, полупродуктов и т.д.) по результатам тестирования проб, взятых из этих упаковок (таблица 1).
Таблица 1- Выбор проб для анализа, исходя из объема партии
| Объем партии (количество упаковок) | Выборка объема пробы на анализ |
| 2-8 | |
| 9-15 | |
| 16-25 | |
| 26-50 | |
| 51-90 | |
| 91-150 | |
| 151-280 | |
| 281-500 | |
| 501-1200 | |
| 1201-3200 | |
| 3201-10000 | |
| 10001-35000 | |
| 35001-150000 | |
| 150001-500000 | |
| 500001 и более |
Приемлемым уровнем качества поступающего сырья, полупродуктов и др. считается получение положительного результата при тестировании всех отобранных проб.
|
|