Главная страница Случайная страница
Разделы сайта
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Мутации,вызывающие наследственные болезни, могут затрагивать структурные, транспортные, эмбриональные белки, ферменты.
|
|
Существует несколько уровней регуляции синтеза белков: претранскрипционный, транскрипционный, трансляционный. Можно предполо-
жить, что на всех этих этапах, осуществляемых соответствующими ферментативными реакциями, могут возникать наследственные аномалии. Если принять, что у человека примерно 80 тыс. генов, а каждый ген может мутировать и обусловливать другое строение белка, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в разных участках гена). На самом деле более чем для 50% белков изменения генетической природы (первичная структура) приводят к гибели клетки и мутация не реализуется в наследственную болезнь. Такие белки называются мономорф-ными. Они обеспечивают основные функции клетки, консервативно сохраняя стабильность видовой организации клетки. Так или иначе, но число генных болезней действительно большое. В настоящее время оно исчисляется тысячами (около 4, 5 тыс.).
При рассмотрении генных болезней как менделирующих форм принимается, что речь идет о так называемых полных формах, т.е. формах, обусловленных гаметическими (в зародышевых клетках) мутациями. Это могут быть новые или унаследованные из предыдущих поколений мутации. Следовательно, в этих случаях патологические гены присутствуют во всех клетках организма. Однако теоретически можно себе представить вероятность появления и мозаичных форм, а не только полных, подобно тому как это уже хорошо известно для хромосомных болезней. Любые мутации, в том числе и генные, могут возникать на ранних стадиях дробления зиготы в одной из клеток, и тогда индивид будет мозаичен по данному гену. В одних клетках у него будет функционировать нормальный аллель, в других - мутантный или патологический. Если эта мутация доминантная, то она проявится в соответствующих клетках и приведет к развитию болезни, очевидно менее тяжелой. Если возникшая мутация в одной из клеток на ранних стадиях развития зародыша рецессивная, то ее эффект проявится только у гомозиготы. Вероятность появления двух новых рецессивных мутаций в одном и том же локусе гомологичных хромосом в одной клетке с двумя нормальными аллелями чрезвычайно мала.
Проблема мозаичных форм генных болезней и с генетической, и с клинической точки
Часть I. ОБЩАЯ НОЗОЛОГИЯ
.
Рис. 23. Родословная семьи с 3 случаями ахондроп-лазии в одном поколении
зрения исследована недостаточно. Частота возникновения мозаичных форм не может быть высокой, поэтому выявлять их трудно. Современные молекулярно-генетические методы позволяют диагностировать мозаицизм на клеточном или тканевом уровне. Обнаружена уже мозаичная форма, легкая по течению, миопатии Дю-шенна (больной умер в возрасте 22 лет).
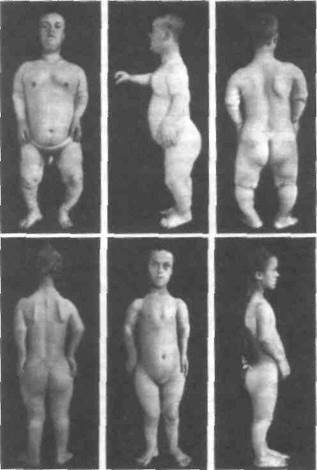
С мозаичными формами генных болезней не следует путать мозаицизм гонад. Мозаицизм гонад является частным случаем органного мо-заицизма, возникающего на более поздних стадиях эмбрионального развития в процессе органогенеза. Наличие его у клинически здорового индивида по какому-либо локусу может обусловить несколько случаев рождения больных детей полной формой наследственной болезни. На рис.23 приведена родословная одной французской семьи с тремя случаями ахондроплазии из четырех детей при здоровых родителях.
Ахондроплазия (рис.24) определенно является аутосомно-доминантным заболеванием с полной пенетрантностью. Клиническая и рентгенологическая диагностика этого заболевания (в частности, в упомянутой выше семье) не вызывает сомнений. Объяснить семейные случаи в данной родословной можно гонадным мозаициз-мом у отца, поскольку больные дети родились в двух его браках. Возможно еще одно объяснение подобных случаев - за счет премутации в одном из аллелей этого гена у родителя, которая реализуется в мутацию при прохождении через мейоз. Однако в гене ахондроплазии премутант-ных состояний пока не обнаружено.
Современными молекулярно-генетическими исследованиями показано, что родительский мозаицизм (в том числе гонадный) ответствен не менее чем за 5-15% случаев доминантных болезней. Мозаицизм у здоровых родителей убедительно доказан при рождении больных детей по соответствующим генам с несовершенным ос-
теогенезом, синдромом Элерса-Данлоса (тип IV), гемофилии (фактор VIII и IX).
Как отмечалось выше, в последние годы обнаружен ряд новых закономерностей наследования признаков (нормальных и патологических), не соответствующих менделевским. К ним относятся: геномный импринтинг и экспансия трип-летных повторов.
|
| Рис. 24. Брат и сестра с ахондроплазией (по W. Falta, 1913) |
Геномный импринтинг представляет собой механизм, с помощью которого различается активность гомологичных генов (или участков хромосом) у индивида в зависимости от их родительского происхождения. У диплоидных организмов обычно экспрессируются оба аллеля какого-то локуса. В участках генома, подверженных импринтингу, экспрессируется только один аллель - отцовский или материнский, т.е. наблюдается моноаллельная экспрессия имприн-
Глава 4 / РОЛЬ НАСЛЕДСТВЕННОСТИ, КОНСТИТУЦИИ И ВОЗРАСТА В ПАТОЛОГИИ
S Зла] Лг S32
тированных генов. Так, известно, что в проксимальном участке хромосомы 15 имеются близко сцепленные, но противоположно импринтирован-ные локусы, отвечающие за возникновение двух фенотипически разных синдромов - Прадера-Вил-ли и Энгельмана. Для синдрома Прадера-Вил-ли, фенотипически проявляющегося умственной отсталостью, мышечной гипотонией, выраженным ожирением, гипогонадизмом, низким ростом и акромикрией, установлен кандидатный ген, ответственный за синтез полипептида N малого ядерного рибонуклеопротеина (SNRPN). Этот ген активно экспрессируется исключительно на отцовской хромосоме 15. Кандидатным геном синдрома Энгельмана (синоним - синдром «счастливой куклы»), характеризующегося неадекватной счастливой улыбкой и глубокой умственной отсталостью с резкими кукольными судорожными движениями, является убиквитин - белковый лигазный ген (ИВЕЗА) Е6-АР, который экспрессируется главным образом на материнской хромосоме 15.
В начале 90-х годов у человека был обнаружен новый тип мутаций, который до сих пор не зарегистрирован ни у одного вида млекопитающих. Он получил название - динамические мутации или мутации экспансии. Суть мутации заключается в нарастании (экспансии) числа триплетных повторов, расположенных в регуля-торной или в кодирующей части генов. К наиболее известным тринуклеотидным повторам, экспансия которых, т.е. увеличение их числа в конкретном участке по сравнению с нормой, приводит к наследственной патологии, относятся повторы CGG (цитозин - гуанин - гуанин) и CAG (цитозин - аденин - гуанин). Увеличение числа повторов данных триплетов от 5-40 в норме до 90-200 приводит к развитию ряда тяжелых неврологических болезней (миотоническая дистрофия, атаксия Фридрейха, синдром ломкой Х-хро-мосомы и др.). Число повторов в норме и при болезнях неодинаково.
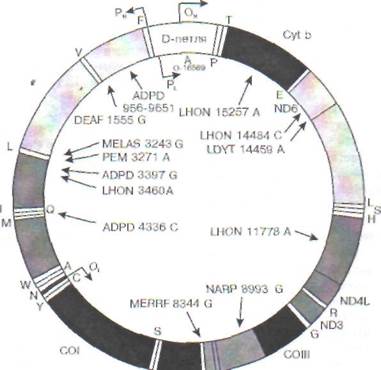
Утверждение, что весь генетический материал человека находится в составе хромосом, не совсем верно, поскольку есть одно исключение - митохондриальный геном. Митохондриальная ДНК (мтДНК) представляет собой небольшую кольцевидную молекулу длиной 16569 пар оснований. В отличие от ДНК ядерного генома она не связана с белками, имеет очень высокую «плотность генов» ввиду отсутствия интронов,
содержит 13 генов, кодирующих белки (3 субъединицы цитохром-с-оксидазы, 6 компонентов АТФазы и др.), 22 гена транспортных РНК и 2 гена рибосомальных РНК. На рис. 25 представлена схема структуры мтДНК и приведены примеры митохондриальных болезней, которые являются следствием мутации мт-генов. Эти болезни передаются только по материнской линии.
Первичные эффекты любых (ядерных и митохондриальных) мутантных аллелей могут проявляться в четырех вариантах: 1) отсутствии синтеза полипептидной цепи (белка); 2) синтезе аномальной по первичной структуре полипептидной цепи (белка); 3) количественно недостаточном синтезе полипептидной цепи (белка); 4) количественно избыточном синтезе полипептидной цепи (белка). На основе первичного эффекта мутантного аллеля (при каждой болезни он всегда один и тот же) уже развертывается весь сложнейший патогенез генной болезни, проявляющийся в разнообразных фенотипических эффектах или клинической картине заболевания.
Результатом действия патологической мутации в онтогенезе (фенотипический эффект) может быть прежде всего летальность на ранних стадиях развития зародыша - до имплантации. Механизмы такой летальности еще не выяснены, но наличие ее у человека не вызывает сомнений. Проявляется это в виде несостоявшегося зачатия (имплантации) у фертильных женщин при нормальной половой жизни. У молодых женщин зачатие наступает в среднем через 3 месяца регулярной половой жизни без контрацепции. Из несостоявшихся зачатий примерно половина обусловлена гибелью зиготы по генетическим причинам (генные, хромосомные и геномные мутации). Если развитие эмбриона с патологической генной мутацией не остановилось на ранних стадиях, то фенотипические эффекты в зависимости от вовлеченного гена и характера мутации формируются в виде трех вариантов: дисморфогенеза (врожденных пороков развития), нарушенного обмена веществ, смешанных эффектов (дисморфогенез и аномальный обмен веществ).
Эффекты патологических мутаций начинают реализовываться в разные периоды онтогенеза: от внутриутробного до пожилого возраста. Большая часть патологических мутаций проявляется внутриутробно (до 25% от общего количества наследственной патологии) и в допубертатном
J4
Часть I. ОБЩАЯ НОЗОЛОГИЯ
возрасте (45%). Еще 20% проявляется в пубертатном и юношеском возрасте и лишь 10% моногенных болезней развивается позже 20 лет.
Важно подчеркнуть, что к одинаковой клинической картине заболевания могут привести нарушения мутации разных генов. Такие случаи называется генокопиями. Наряду с этим, хотя и редко, могут встречаться фенокопии генных болезней. Это те случаи, при которых повреждающие внешние факторы, действующие, как правило, внутриутробно, вызывают болезнь, сходную по клинической картине с наследственной. Противоположное состояние, когда при мутантном генотипе индивида в результате сре-довых воздействий (лекарства, диета и т.п.) болезнь не развивается, называют нормокопиро-ванием. Понятия о гено- и фенокопиях помогают врачу поставить правильный диагноз, а также более точно определить возможность возникновения заболевания и вероятность рождения больного ребенка. Понимание принципов нормо-копирования дает врачу возможность вторичной профилактики наследственных болезней, а в каждом конкретном случае предупредить развитие болезни у ребенка, унаследовавшего патологический ген.
Патогенез генных болезнейсвязан с первичным эффектом мутантного аллеля. Поэтому принципиальные позиции патогенеза генных болезнейможно представить следующим образом: мутантный аллель -► патологический первичный продукт (качественно или количественно) -> цепь последующих биохимических процессов -> клетки -» органы -> организм.
Это и есть главная общая закономерность развития генных болезней при всем их многообразии. В зависимости от того, какой продукт контролируется конкретным геном и каков характер нарушения его при мутации, соответствующим образом развертывается патогенез болезни на молекулярном уровне.
Если в результате мутации вырабатывается избыточное количество белка, то весь патогенез болезни в целом будет обусловлен именно гиперпродукцией генной активности. Так, при первичном гемохроматозе синтезируется избыточное количество глобина, что приводит к пе-ренагруженности эритроцитов гемоглобином и, соответственно, железом. Увеличивается свертываемость крови, развивается гемосидероз паренхиматозных органов.
При другом варианте патологического эффекта
|
| 12s rRNA |
| 16s rRNA |
| ND5 |
| ND1 ND2 |
| ND4 |
D СОН К ATPase6
Рис. 25. Структура митохондриально-
го генома и примеры митохондриаль-
ных болезней:
ADPD - болезнь Альцгеймера/болезнь
Паркинсона;
DEAF - нейросенсорная потеря слуха;
LHON - наследственная нейроофталь-
мопатия Лебера;
LDYT - LHON и дистония;
MELAS - митохондриальная миопа-
тия, энцефалопатия, молочнокислый
ацидоз и приступы судорог;
MERRF - миоклональная эпилепсия в
сочетании с необычно красными
мышечными волокнами;
NARP - нейропатия, атаксия и пигментный ретинит;
РЕМ - летальная прогрессирующая
энцефаломиопатия
Глава 4 / РОЛЬ НАСЛЕДСТВЕННОСТИ, КОНСТИТУЦИИ И ВОЗРАСТА В ПАТОЛОГИИ
мутантного гена вырабатывается аномальный белок. За этим следуют фукциональные нарушения в системе, которую в норме обеспечивает нормальный белок. Нарушения эти первоначально развертываются на молекулярном уровне. Примером такого варианта патогенеза болезни может быть серповидно-клеточная анемия. Замена гидрофильной глютаминовой кислоты на гидрофобный валин в структуре глобина изменяет функциональные свойства гемоглобина (пониженная растворимость, повышенная полимеризация). Он не может выполнять кислород-акцепторную функцию и кристаллизуется при недостатке кислорода. Эритроциты приобретают поэтому серповидную форму (отсюда и название болезни), склеиваются и тромбируют капилляры и т.п.
Третий вариант патологического эффекта мутантного аллеля - отсутствие выработки первичного продукта. Это выражается в накоплении токсических продуктов-предшественников. Например, при фенилкетонурии накапливается в крови фенилаланин, поскольку он из-за отсутствия фенилаланингидроксилазы печени не превращается в тирозин. Могут использоваться другие (обходные) пути обмена, часто также с патологическим исходом. В результате отсутствия первичного продукта гена может задерживаться какой-либо важный процесс, постоянно осуществляющийся в организме. Так, мутации генов, детерминирующих ферменты репарации ДНК, делают невозможным восстановление постоянно возникающих нарушений в структуре ДНК, что приводит к развитию злокачественных новообразований (пигментная ксеродерма, атак-сия-телеангиэктазия).
Известен и четвертый вариант первичного патологического эффекта мутантного аллеля - это выработка уменьшенного количества нормального первичного продукта (талассемия, аката-лаземия). Патогенез таких заболеваний отличается большой вариабельностью, поскольку наряду с нормальным путем обмена веществ будут функционировать и патологические варианты.
Выше были описаны общие закономерности патогенеза генных болезней на молекулярном уровне на примерах нарушения обмена веществ. Тот же самый принцип патогенеза (мутантный аллель - патологический первичный продукт) действует и для генов морфогенетического контроля, мутации в которых приводят к врожден-
ным порокам развития (полидактилия, синдромы Холт-Орама, Нунена, Лоуренса-Муна, Мек-келя, Робертса). Начальное звено врожденного порока развития связано с нарушением диффе-ренцировки клеток. Запрограммированная в геноме дифференцировка клеток, а затем и органогенез осуществляются путем смены активации и выключения определенных генов в строго ограниченных временных (по отношению к онтогенезу) промежутках. Если первичный продукт морфогенетического гена аномальный, то не последует необходимая для дальнейшего правильного развития органа дифференцировка клеток. Естественно, что морфогенетических генов много, действуют они в разные периоды онтогенеза. Соответственно, мутации в них будут приводить к специфическим врожденным порокам развития.
Патогенез генных болезней не заканчивается на молекулярном уровне даже в его первичных звеньях. Для многих болезней основным звеном патогенеза является клеточный уровень. При этом речь идет не только о биологической аксиоме: во всех генетических процессах клетка является дискретной самостоятельно регулируемой единицей и в ней осуществляются все процессы реализации генетической информации (транскрипция, трансляция, синтез белка). Клеточный уровень патогенеза генных болезней означает, что в определенных типах клеток разыгрываются основные патологические процессы, характерные для конкретной нозологической формы. Клетка как бы «не выпускает из себя» патологические явления, а берет удар от первичного патологического эффекта гена на себя. Точкой приложения являются отдельные структуры клетки, разные при разных болезнях (лизосо-мы, пероксисомы, мембраны).
Патогенез на клеточном уровне развертывается при болезнях накопления в связи с нарушением ферментативной активности в лизосо-мах. Так, накопление в клетках гликозоами-ногликанов (мукополисахаридов), а в последующем и в основном межклеточном веществе приводит к развитию тяжелой группы заболеваний
- мукополисахаридозов. Причиной избыточного содержания полимеров - гликозоамингликанов
- является отсутствие их деградации в лизосо-мах (рис.26, вклейка). Нарушение распада гликозоамингликанов связано с дефектами в группе специфических ферментов, осуществляющих весь цикл деградации.
Часть I. ОБЩАЯ НОЗОЛОГИЯ
Эффекты мутаций в гене рецептора ЛНП на генный продукт
Таблица 13
| Класс мутаций | Последствия |
| Нулевой | Нетбелка-рецептора |
| Дефект транспорта поверхности | Отсутствуют или уменьшено число рецепторов на клеточной |
| Дефект связывания | Нормальное число рецепторов, но отсутствует или уменьшено связывание ЛНП |
| Дефект интернализации | Нормальное число рецепторов и связывание ЛНП; отсутствие или снижение эндоцитоза ЛНП |
| Дефект возвращения | Нормальное число рецепторов, связывание ЛНП и эндоцитоз; отсутствует или уменьшено освобождение ЛНП и возвращение рецептора на клеточную поверхность |
Другим примером болезней накопления могут быть гликогенозы. В клетках печени и мышц накапливаются полимеры гликогена, которые не подвергаются деградации даже тогда, когда организму необходима глюкоза в крови. Принципиально патогенез гликогенозов такой же, как и мукополисахаридозов. В клетках печени и мышц отсутствует определенный фермент (их уже известно много), который участвует в цикле расщепления гликогена до глюкозы.
Другие внутриклеточные структуры - перок-сисомы - также могут являться точкой приложения первичного действия мутантного гена. В этих случаях развиваются так называемые перок-сисомные болезни. К этой группе относятся синдром Цельвегера, ризомелическая точечная ос-теохондродисплазия, болезнь Рефсума, акатала-зия и др.
Мембраны клеток также могут быть ключевыми элементами патогенеза генных болезней. Так, нарушение синтеза рецепторов андрогенов приводит при хромосомном наборе ХУ к развитию женского фенотипа, наличию семенников в брюшной полости и повышенному уровню андрогенов (синдром тестикулярной феминизации). Клиника витамин Д-резистентного рахита (ауто-сомно-доминантное заболевание) обусловлена дефектом рецепторов 1, 25-дигидроксихолекаль-циферолла. Патологические мутации в гене рецептора липопротеидов низкой плотности вызывает пять видов дефектов клеточного рецептора. Эффект любой мутации - нарушение гомеостаза холестеринового обмена (табл. 13).
При муковисцидозе (синоним - кистозный фиброз) нарушается регуляция транспорта хлоридов через мембраны эпителиальных клеток, которая в норме осуществляется белком - продук-
том гена, названным «кистофиброзным трансмембранным регулятором» (сокращенно CFTR). Одни мутации в гене CFTR ведут к снижению синтеза белка CFTR из-за незавершенности процессинга РНК, другие - к качественным изменениям мембранных хлоридных каналов. Одна первичная биохимическая аномалия (нарушение транспорта хлоридов) ведет к мультиорганному патологическому процессу (прогрессирующее поражение дыхательных путей, хронические синуситы, недостаточность экзогенной секреторной функции поджелудочной железы, стерильность у мужчин).
Клеточный уровень патогенеза генных болезней может проявляться не только в конкретных органеллах, но и в виде нарушения скоординированное™ функций интерфазного периода клетки и размножения. Так, мутации, затрагивающие области онкогенов, ведут к снятию контроля размножения клеток (репрессия антионкогенов) и, соответственно, к злокачественному росту (наследственные раки толстого кишечника, ретинобл астома).
Клетка может быть главным звеном при реализации молекулярного уровня патогенеза. Так, прекращение синтеза мышечного белка дистро-фина при мутациях в этом гене приводит к постепенной деградации мышечных клеток. Это и является основным звеном патогенеза тяжелой наследственной болезни - миопатии Дюшенна.
Органный уровень патогенеза наследственных болезней, безусловно, является производным от молекулярного и клеточного. При различных болезнях мишенью патологического процесса служат разные органы, иногда в результате первичных процессов, иногда - вторичных. Так, отложение меди в печени и в экстра-
Глава 4 / РОЛЬ НАСЛЕДСТВЕННОСТИ, КОНСТИТУЦИИ И ВОЗРАСТА В ПАТОЛОГИИ
пирамидной системе мозга при гепато-лентику-лярной дегенерации (болезнь Вильсона) является первичным процессом, а гемосидероз паренхиматозных органов при первичном гемохрома-тозе или талассемии развивается вторично вследствие усиленного распада эритроцитов. При ал-каптонурии отложения гомогентизиновой кислоты в хрящах суставных поверхностей и клапанах сердца являются вторичным процессом, обусловленным высокой концентрацией гомогентизиновой кислоты в крови (она не превращается в малеилацетоуксусную кислоту в результате мутационно обусловленного отсутствия оксида-зы гомогентизиновой кислоты). Это медленно ведет (примерно к 40 годам) к порокам сердца и тугоподвижности суставов.
В организме в целом взаимосвязь патогенетических процессов проявляется сочетанно на молекулярном, клеточном и органном уровнях. Патологический процесс, индуцированный первичным эффектом мутантного аллеля, приобретает целостность с закономерными межиндивидуальными вариациями. Тяжесть и скорость развития болезни при прочих равных условиях (пол ребенка, одинаковый характер мутации) зависят от генотипа организма (гены-модификаторы) и условий среды. Патогенез любой наследственной болезни у разных индивидов, хотя и сходен по первичным механизмам и этапам, формируется строго индивидуально.
4.1.8. Этиология и патогенез
хромосомных болезней
|
|